MDS Kategorien nach WHO 
•
Refraktäre Anämie
o
ohne Ringsideroblasten (RA)
o
mit Ringsideroblasten (RARS)
•
Refraktäre Zytopenie mit multipler Dysplasie (RCMD)
•
Refraktäre Sideroblastische Zytopenie mit multilinearer Dysplasie (RSCMD)
•
Refraktäre Anämie mit vermehrten Blasten
o
mit 5-10 % Blasten (RAEB I)
o
mit 11-20 % Blasten (RAEB II)
•
5 q Syndrom
•
unklassifiziert
Tab 1: Klassifikation der MDS (69)
Obwohl zahlreiche genetische und morphologische Aberrationen bei diesen Erkrankungen vorkommen (z.B.
zelluläre Atypien und chromosomale Abnormalitäten in 50% der Fälle, wie Trisomie 8, Chromosom 5 & 7
Deletionen), ist die Ursache der meisten Fälle von MDS bisher unbekannt. Die genetischen Defekte liegen
bereits in den hämatopoetischen Stammzellen (65). Als sekundäre Formen von MDS werden die Fälle
angesehen, die nach chronischer oder Hochdosis-Exposition mit alkylierenden Substanzen, bestimmten
Umweltgiften oder radioaktiver Strahlung auftreten.
Prognose und Therapie bei MDS
MDS ist eine Ausschlussdiagnose, basierend auf der Existenz einer persistierenden (> 6 Monate) Zytopenie
einer oder mehrerer Zelllinien bei gleichzeitigen Nachweis von Dysplasien in einer oder mehreren
hämatopoetischen Zelllinien im Knochenmark mit oder ohne erhöhte Blastenzahl. Die große Heterogenität der
Krankheit bedingt sehr große Unterschiede in der Überlebensdauer zwischen einzelnen Patienten. Seit 1997
wird das International Prognostic Scoring System (IPSS) (70) verwendet (Tabelle 2).
Prognostische Variable Punkte
0
0.5
1.0
1.5
2
KM Blasten
<5 %
5-10 %
-
11-20 %
21-30 %
Karyotyp1
Gut
Mittel
Schlecht
Cytopenie2
0-1
2-3
Risiko Score
Risiko
0
0.5-1
1.5-2
>2.5
Niedrig
Mittel-1
Mittel-2
hoch
Vorausgesagte mediane Überlebensdauer
5.7 J
3.5 J
1.2 J
4.8 Monate
1Gut = normal, -Y, del(5q), del(20q); schlecht = komplex (= 3 Abnormalitäten) oder Chromosom 7
Abnormalität; and mittel = andere karyotypische Abnormalität
2 Neutrophile <1.8 x 109/L, Hämoglobin <10 g/dL, Thrombozyten <100 x 109/L
Tab.2: Internationales Scoring-System zur Einschätzung der Prognose bei Myelodysplastischem Syndrom
(International Prognostic Scoring System (IPSS) (modifiziert nach 70)
Für den einzelnen Patienten hängt die Prognose vom individuell vorliegenden Subtyp und vom IPSS Score ab
(Tabelle 3.5). Patienten mit RA, RARS und niedrigem IPSS entwickeln selten eine AML und können viele Jahre
mit einem MDS leben. Hochrisiko-Patienten mit RAEB1, RAEB2 und hohem IPSS Score zeigen einen
progressiven Krankheitsverlauf und benötigen gegebenenfalls eine aggressivere Therapieform. Die meisten
Patienten versterben durch die sich entwickelnden Leukämien oder als Folge des Knochenmarksversagens
durch unbeherrschbare Infektionen oder Blutungskomplikationen.
Die MDS sollten wegen ihrer Vielgestaltigkeit und dem meist höheren Lebensalter der Patienten möglichst
risikoadaptiert individuell angepasst therapiert werden (71,72). Die Therapiestrategien bewegen sich je nach
individuellem Risiko-Profil des Patienten zwischen rein supportiven Maßnahmen und Therapien für
Hochrisikopatienten wie Chemotherapie/Stammzelltransplantation und Immunsuppression mit ALG/ATG bei
Niedrigrisiko MDS.
Die hämatopoetische Stammzelltransplantation (HSCT) eröffnet die einzige kurative Heilungschance bei MDS.
Dies ist aber auch zugleich die Behandlungsform mit der höchsten Lethalität. Außerdem sind die besten Erfolge
bei jungen Patienten mit den risikoärmsten Formen (RA und RARS, normale Zytogenetik) zu beobachten, die
auch bei einer konservativen Therapie am besten abschneiden. In einer retrospektiven Auswertung von 184
europäischen Patienten, die mit autologer Stammzelltransplantation behandelt wurden, ergab sich bei 26 %
eine 4-Jahre-krankheitsfreie Überlebensrate (73). Ein Vorteil gegenüber einer allogenen Transplantation ist
offenbar nicht gegeben. Aufgrund des höheren Alters der meisten Patienten bei Diagnosestellung kommt die
HSCT bei den meisten Patienten mit MDS allerdings nicht in Betracht.
Eine intensive Chemotherapie ist risikoreich und von wechselndem Erfolg. 30% der MDS Patienten mit
niedrigem Risiko sprechen auf eine immunsuppressive Therapie mit ATG (Anti-Thymozyten-Globulin) mit einer
stabilen Remission an.
Die supportive Therapie mit Erythrozyten/Thombozytenkonzentraten und mit Breitbandantibiotika ist die
Therapieform für die meisten MDS-Patienten. Klinisch ist die ineffektive Erythropoese von großer Bedeutung.
Eine Anämie ist in 80-90 % der Patienten vorhanden. Müdigkeit, Schwäche, Atemnot sind deshalb häufig auch
die ersten Symptome bei sich entwickelnder MDS. Mehr als 40% der MDS-Patienten brauchen im Laufe der
Erkrankung Bluttransfusionen, eine Reihe werden auf Dauer transfusionspflichtig (74). Bei chronischer
Transfusionstherapie ist eine sich entwickelnde Eisenüberladung durch die Transfusionen unvermeidlich.
Klinische Konsequenzen der Eisenüberladung bei MDS
Nach den bereits etwas älteren Arbeiten von Jensen aus Dänemark reduziert die Therapie mit dem Chelator
Deferoxamin (DFO) die Anzahl der notwendigen Transfusionen bei MDS (75,76). Aktuell wird mit neuem
Interesse diskutiert, welche Bedeutung eine sich entwickelnde Eisenüberladung bei MDS hat und welche
Bedeutung einer optimierten Eisenentzugstherapie bei MDS-Patienten zukommt.
Im Mai 2005 wurde dazu ein Consensus Meeting „Iron overload in myelodysplastic syndrome“ in
Nagasaki/Japan abgehalten, auf der führende Experten Fragen nach der Diagnose und Behandlung von
Eisenüberladung bei MDS diskutiert haben (76). Gemeinsam wurde festgestellt:
• Die klinische Konsequenz von nicht- oder ungenügend behandelter Eisenüberladung bei MDS sind
mögliche kardiale-, hepatische oder endokrine Komplikationen.
• Ziel der Eisenchelatorbehandlung bei MDS sind: Vermeidung und Behandlung von diesen
eiseninduzierten Komplikationen. Steigerung der Lebenserwartung.
• Die Chelattherapie ist möglicherweise klinisch besonders wichtig in einer Untergruppe von MDS-
Patienten
Der Grad der Eisenüberladung sollte bei Diagnosestellung und in regelmäßigen Intervallen danach in
Anhängigkeit von der Transfusionsfrequenz untersucht werden (76). Dies trägt der Tatsache Rechnung, dass
einige MDS-Patienten bereits zum Zeitpunkt der Diagnosestellung eisenüberladen sind, wahrscheinlich bedingt
durch erhöhte Nahrungseisenabsorption infolge der ineffektiven Erythropoese und der Anämie. Als
diagnostische Parameter kommt in erster Linie das einfach zu messende Serum-Ferritin in Betracht (alle 3
Monate). Falls nichtinvasive Methoden zur Lebereisenquantifizierung wie Magnetresonanztomographie oder
SQUID-Biosuszeptometrie zur Verfügung stehen, sollte alle 12 Monate eine Messung erfolgen. Wiederholte
Leberbiopsien sind wegen des Blutungsrisikos bei MDS-Patienten eher abzulehnen.
Die Entscheidung für den Einsatz einer Eisenchelattherapie sollte in Abhängigkeit von der Zahl der
Transfusionen und dem Grad der individuell vorliegenden Eisenüberladung (Ferritin, Lebereisen) getroffen
werden. Allgemein wird ein Ferritinwert von > 1000-2000 µ/l angegeben, wobei die aktuelle
Transfusionsfrequenz wichtiger ist als die absolute Zahl der Transfusionen. Die Eisenchelattherapie sollte
fortgesetzt werden, solange die Transfusionstherapie anhält.
Von einer Eisenchelattherapie werden vor allem Patienten mit hohem Transfusionsbedarf und vorhandener
Eisenüberladung profitieren. In Fällen mit individuell schlechter Prognose macht eine Chelattherapie dennoch
Sinn, wenn typische eiseninduzierte Organschäden vorliegen, wie z.B. eine Myokardsiderose und ihre Folgen.
Allgemein ist wahrscheinlich ein Nutzen bei Patienten mit IPSS low oder intermediate-1 und gemäß WHO
Klassifikation RA, RARS sowie 5q-Syndrom zu erwarten.
Außerdem sollten unabhängig vom IPSS-Score alle Kandidaten für eine KMT als Chelator-Patienten eingestuft
werden, weil eine Chelattherapie die Bildung von eiseninduzierten Organschäden verhindert und eine optimale
Organfunktion mitentscheidend für den Erfolg einer Transplantation ist. Momentan sind noch viele Fragen über
den Einsatz von Eisenchelatoren bei MDS offen. Standardtherapie ist momentan der Einsatz von Deferoxamin,
wobei die subkutane Infusion für die älteren Patienten sicher unbequem und belastend ist. Der orale Chelator
Deferipron ist in Europa bisher nicht zugelassen für MDS-Patienten. Studien mit dem neuen Chelator
Deferasirox wurden und werden z.Zt. durchgeführt (77)
Literatur
64. Greenberg PL , Young NS, Gattermann N. Myelodysplastic Syndromes. Hematology (Am Soc Hematol
Educ Program) 2002; 136-161
65 Bowen D, Culligan D, Jowitt S, Kelsey S, Mufti G, Oscier D, Parker J. Guidelines for the diagnosis and
therapy of adult myelodysplastic syndromes. Br J Haematol 2003; 20:187-200.
66 Hofmann WK, Koeffler HP. Myelodysplastic syndrome. Annu Rev Med 2005;56:1-16.
67 Germing U, Strupp C, Kundgen A, Bowen D, Aul C, Haas R, Gattermann N. No increase in age-specific
incidence of myelodysplastic syndromes. Haematologica. 2004; 89(8): 905-10
68 Harris N, Jaffe E, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, Lister TA, Bloomfield C. WHO
Classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the clinical advisory
committee meeting. J Clin Oncol 1999; 17:3835-3849
69. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in
myelodysplastic syndromes. Blood 1997; 89:2079-88. [Erratum, Blood 1998; 91:1100]
70. Alessandrino EP, Amadori S, Barosi G, et al. Evidence- and consensusbased practice guidelines for the
therapy of primary myelodysplastic syndromes. A statement from the Italian Society of Hematology.
Hematologica 2002; 87:1286-1306
71. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Myelodysplastic
syndromes. version 1.2005. Accessed August 10, 2005 (http://www.nccn.org).
72. de Witte T, Suciu S, Verhoef G, et al: Intensive chemotherapy followed by allogeneic or autologous
stem cell transplantation for patients with myelodysplastic syndromes (MDSs) and acute myeloid leukemia
following MDS. Blood 2001; 98:2326-2331.
73 Cazzola M, Malcovati L. Myelodysplastic syndromes—coping with ineffective hematopoiesis. N Engl J
Med 2005; 352:536-538
74 Jensen PD, Jensen IM, Ellegaard J. Desferrioxamine treatment reduces blood transfusion requirements
in patients with myelodysplastic syndrome. British Journal of Haematology 1992; 80(1):121-124
75. Jensen PD, Heickendorff L, Pedersen B, Bendix-Hansen K, Jensen FT, Christensen T, Boesen AM,
Ellegaard J.The effect of iron chelation on haemopoiesis in MDS patients with transfusional iron overload. Br J
Haematol. 1996; 94(2):288-99
76. Gattermann N, Porter J, Lopes LF, Seymour J Consensus statement on iron overload in myelodysplastic
syndromes Clinical Cornerstone. Hematolgy/Oncology Clinics 19, Supplement 1. July 2005; 18-25
77. Cazzola M, Gattermann N, Greenberg P, Maertens J, Soulieres D, Rose D, Ressayre Djaffer C, Rabault
B, Ford JM, Alberti D. ICL670, a once-daily oral iron chelator, is effective and well tolerated in patients
with myelodysplastic syndrome (MDS) and iron overload. Leuk Res 2005; 29 (Suppl.1):S67















© www.eiseninfo.de
Myelodysplastisches Syndrom
(MDS)
Die myelodysplastischen Syndrome (MDS) bilden eine
heterogene Gruppe von Erkrankungen des myeloischen
Systems, die charakterisiert sind durch eine
hämatopoetische Insuffizienz verbunden mit einer klinisch
relevanten Zytopenie und dem zusätzlichen Risiko (ca.
10-40 %) für eine maligne Transformation in Richtung
einer akuten myeloischen Leukämie (AML) (64-67).
Die MDS zählt zu den häufigsten hämatologischen
Erkrankungen überhaupt, wobei die Inzidenz in den
letzten 30 Jahren wohl nicht angestiegen ist (68).
Betroffen sind überwiegend ältere Menschen, ca. 20
bis 50 pro 100,000 Personen erkranken jedes Jahr
in unserer Bevölkerung. 90 % der Patienten sind
älter als 50 Jahre (Median 65-75 Jahre), bis zu 5 %
der Anämien bei älteren Patienten sind bedingt
durch ein MDS. Tabelle 1 zeigt eine Klassifizierung
der MDS nach den WHO- Kriterien (69).

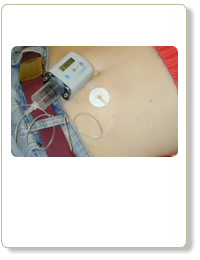
Eisenüberladung
Subkutane Injektion von DesferalR, einem Eisenchelator, der Eisen im Körper mobilisieren und ausscheiden lassen kann