Wie wichtig Eisen im medizinischen Sinne ist, ergibt sich aus der Tatsache, dass Erkrankungen, die mit einem
Mangel oder einer Überladung von Eisen verbunden sind, weltweit außerordentlich häufig vorkommen. Nach
WHO-Angaben leiden 30 % der Weltbevölkerung an einer Eisenmangelanämie (1). Wenn man unter den
Eisenüberladungserkrankungen nur die hereditäre Hämochromatose betrachtet, so gilt allein diese als die
häufigste monogen bedingte Erbkrankheit in der westlichen Welt. Jeder 200.-300. Europäer, Amerikaner und
Australier ist davon homozygot betroffen (2).
Historisch gesehen war die Eisenmangelanämie bei jungen Frauen als Morbus Virginum, später als Chlorosis seit dem
17. Jahrhundert bekannt. 1936 fand Heilmeyer den Zusammenhang dieser Anämieform mit niedrigem Serum-Eisen (3).
Viele Jahrzehnte galt die Physiologie des Eisenstoffwechsels als fast rätselhaft komplex. Molekularbiologische Methoden
haben in den letzten 10 Jahren neue Erkenntnisse erbracht, sodass heute die intestinale Eisenabsorption, der Transport
von Eisen in Zellen und aus Zellen heraus, der intrazelluläre Eisenstoffwechsel im Wesentlichen als verstanden gelten
können.
Die „neue Zeitrechnung“ in der Eisenstoffwechselforschung begann mit der Suche nach der Ursache der erblichen
Eisenspeicherkrankheit (hereditäre Hämochromatose). 1996 wurde die C282Y-Mutation im HFE-Gen nachgewiesen und
damit das Gen und das Genprodukt (HFE-Protein) gefunden (4). Seither sind viele Rezeptoren, Transportwege und
Regulationsmechanismen bekannt geworden.
Viele Störungen des Eisenstoffwechsels beruhen auf genetischen Veränderungen von beteiligten Proteinen, sodass die
Kenntnis dieser Zusammenhänge wesentlich zum tieferen Verständnis der Pathophysiologie, Diagnostik und Therapie von
Eisenstoffwechselkrankheiten geführt hat. Dies gilt insbesondere für die Eisenüberladungserkrankungen.
Die Bedeutung von Eisen im Sauerstofftransportprotein Hämoglobin ist lange bekannt. Des Weiteren sind eisenabhängige
Enzyme (Oxidoreduktasen, Monooxygenasen, Dioxygenasen, 2Fe-2S, 4Fe-4S-Enzyme) an allen wichtigen
Stoffwechselzyklen beteiligt. So finden sich in der inneren Membran von Mitochondrien Cytochrome, die wesentliche
Funktionen bei der oxidativen Phosphorylierung einnehmen. Zu den eisenhaltigen Oxidoreduktasen gehört z.B. die
Ribonukleotidreduktase, das Schlüsselenzym der DNA-Synthese. Die Cytochrom P450-Familie katalysiert hunderte von
Reaktionen im Fremdstoffmetabolismus. Fettsäuredesaturasen, Lipoxygenasen, Peroxidasen, NO-Synthetasen, die
Akonitase im Citratcyclus, die Guanylatcyclase (Signaltransduktion, second messenger) und die
Aminophosporibosyltransferase (Purinsynthese) sind gleichfalls eisenhaltige Enzyme (5, 6).
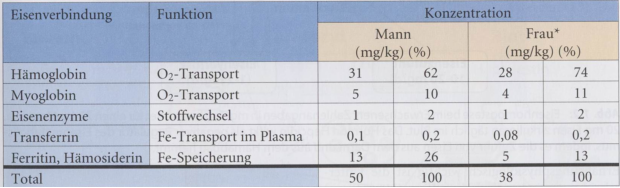
Der menschliche Körper eines Erwachsenen enthält 3-5 g Eisen hauptsächlich in Form von Hämoglobin, als Häm- oder
nicht-Häm-Eisen-Enzymen und als Depot-Eisen, gespeichert in Ferritin und Hämosiderin (Tabelle 1). Der tägliche
physiologische Eisenverlust in Form von abgeschilferten Epithelzellen der äußeren und inneren Körperoberflächen, sowie
durch Schweiß und Urin beträgt insgesamt ca. 1-2 mg und ist nicht regulierbar.
Tab. 1: Verteilung von Eisen im Körper
Dieser Verlust wird durch die Eisenaufnahme mit der Nahrung normalerweise genau ausgeglichen. Dabei wird auch die
Speicherung von Reserveeisen in Form von Ferritin und Hämosiderin in Leber, Milz, Knochenmark und Muskulatur
aufrechterhalten. Beim erwachsenen Mann sind dies ca. 800 mg, bei Frauen und Kindern deutlich weniger. Die täglich im
Körper zirkulierende Eisenmasse ist wesentlich größer als der Eisenverlust oder die Eisenaufnahme (Abb. 1).
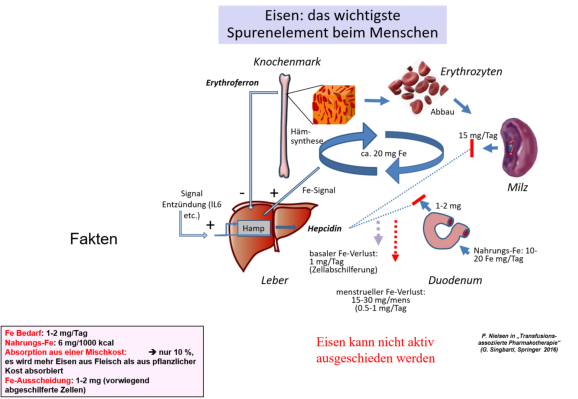
Abb. 1: Eisenhomöostase beim Erwachsenen. Zahlenangaben in mg/Tag berechnet für einen 70-kg Mann. Ca. 20 mg Eisen
zirkulieren täglich im Blut. Das Hormon Hepcidin wirkt als negativer Regulator des Eisenmetabolismus, indem es die Zufuhr von Eisen
aus dem Darm und aus dem Hämabbau hemmt (aus Nielsen Diagnostik und Therapie von Eisenmangel mit und ohne Anämie, Uni-
Med. 2 Auflage 2016).
Intestinale EisenabsorptionEisen ist in der Ökosphäre ubiquitär, kommt aber in Nahrung und Trinkwasser nur in geringen Konzentrationen vor.
Komplizierte Aufnahmemechanismen im Darm sind nötig, um dem Bedarf angepasste Eisenmassen (normal 1-2 mg/Tag,
maximal 3-5 mg/Tag) aufzunehmen.
Ernährungsphysiologisch wichtig ist die Unterscheidung zwischen tierischem Eisen (Häm-Eisen) und pflanzlichen
Nahrungseisen (nicht-Häm-Eisen). Der Hauptort für die Absorption von Eisen in allen Formen sind das Duodenum und
das obere Jejunum. Im starken Eisenmangel können aber auch tiefere Darmabschnitte zusätzlich substantielle
Eisenmengen aufnehmen.
Pflanzliches Eisen in der Nahrung liegt größtenteils als polymerer Fe(III)-hydroxid-Kohlenhydrat-Komplex vor. Es wird
daraus im sauren Magenmilieu, besonders gut unter reduzierenden Bedingungen (Vitamin C), herausgelöst und kann als
zweiwertiges ionisches Eisen unter den Bedingungen im Duodenum (Übergang saurer-neutraler pH-Wert) gut löslich
gehalten und dann absorbiert werden. Dreiwertiges ionisches Eisen unterliegt bei neutralem pH schnell der
Hydroxylierung, wobei extrem schwerlösliches Eisen(III)-Hydroxid entsteht, was nicht mehr aufgenommen werden kann.
Bei der Reduktion von pflanzlichem Nahrungseisen spielt möglicherweise die cytochromhaltige Eisenreduktase Dcytb in
der Bürstensaummembran des Enterozyten eine Rolle (Abb. 2) (7).
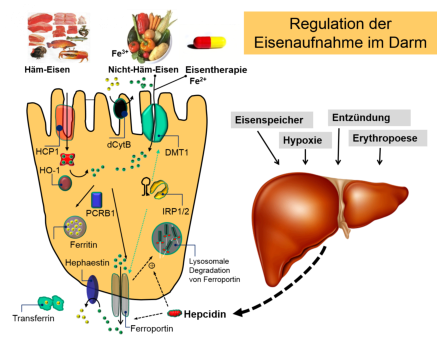
Abb. 2 Regulation der intestinalen Eisenabsorption von tierischem und pflanzlichem Eisen. Modifiziert nach P. Nielsen „Eisen-
Pharmakotherapie von Eisenmangel und Eisenüberladung „in „Allgemeine und spezielle Pharmakologie & Toxikologie„ 11. Aufl. 2013,
Elsevier
Lösliches, ionisches Fe(II) wird beim Menschen über den divalenten Metall-Ionen-Transporter (DMT1) in den Enterozyten
aufgenommen (8). Dieses Protein bewirkt einen Protonen-vermittelten Kationentransport für Fe2+, Zn2+, Mn2+, Co2+,
Cd2+, Cu2+, Ni2+, und Pb2+. Beim Menschen ist DMT1 offenbar vorwiegend für den Transport von Eisen(II) zuständig
(9). Die DMT1-Expression im Duodenum ist im Eisenmangel erhöht, Mutationen in DMT1 bewirken einen systemischen
Eisenmangel mit Anämie. Offenbar gibt es beim Menschen verschiedene DMT1 Varianten mit und ohne 3’ IRE oder 5’-IRE
-Aktivität, die eine komplexe Regulation der Eisenaufnahme in verschiedenen Geweben bewerkstelligen können (10).
Wenn Eisen im Organismus benötigt wird, dann erfolgt der Transport aus dem Enterozyten ins Pfortaderblut sehr rasch.
Der basolaterale Transfer wird durch das Membranprotein IREG1 (in USA auch Ferroportin1 genannt) bewerkstelligt (11,
12).
Das aus dem Enterozyten ausgeschleuste zweiwertige Eisen wird über die kupferhaltige Ferroxidase Hephaestin oxidiert,
um dann im Pfordaderblut an Apotransferrin gebunden zu werden (13). Unklar ist weiterhin die Rolle von HFE
(Genprodukt des Hämochromatose-Gens) an der Regulation der intestinalen Eisenabsorption.
Ergänzend wird in der Literatur von einer Arbeitsgruppe ein zusätzlicher Aufnahmemechanismus von Fe(III) (Ferri-Eisen)
im Darm über den „IMP-Weg” (Integrin/Mobilferrin/Paraferritin) postuliert (14). Versuche mit blockierenden Antikörpern
und konkurrierenden Kationen (speziell Mn2+) legen nahe, dass beide Aufnahmewege (Fe2+, Fe3+) unabhängig
voneinander sind. Nach gegenwärtigem Stand wird von den meisten Autoren in diesem Gebiet jedoch bezweifelt, dass
beim Menschen die Absorption von Fe(III) nennenswert zur Versorgung mit Nahrungs-Eisen beiträgt, ebenso ist die
Bioverfügbarkeit von oralen Eisen(III)-Verbindungen in der Eisentherapie nur sehr gering.
Aus einer typischen westlichen Mischkosternährung mit 6 mg Fe/1000 kcal wird ca. 1/3 der täglichen
Nahrungseisenaufnahme aus Häm-Eisen gedeckt, obgleich nur 10-15 % des Nahrungseisens aus Häm-Fe bestehen (15).
Häm-Fe aus Fleisch wird also sehr effizient über spezifische, mukosale Bürstensaum-Rezeptoren aufgenommen. Ein
daran beteiligtes Protein (heme-carrier-protein 1, HCP1) ist vor kurzem gefunden worden (16). Zuerst wird im Darmlumen
Häm aus Hämo- bzw. Myoglobin freigesetzt und auf unbekannte Weise stabilisiert, Häm wird dann als intaktes Molekül
aufgenommen. Der geschwindigkeitsbestimmende Schritt in der Häm-Fe-Absorption ist der Abbau von Häm durch eine
Hämoxygenase im Enterozyten (17) (Abb. 2.). Hemmstoffe der Hämoxigenase inhibieren die Häm-Eisen-Absorption (18).
Da die Applikation von hohen Dosen Häm-Fe auch die Absorption von nicht-Häm-Fe und umgekehrt hemmt, muss das
aus Häm-Fe freigesetzte Eisen in den gleichen intrazellulären Eisenpool eingespeist werden wie Eisen aus der nicht-
Häm-Fe-Absorption. Dies gilt auch für den Abtransport aus den Enterozyten über IREG1/Ferroportin1, der für alle
absorbierten, ehemals unterschiedlichen Eisenverbindungen (Häm-Fe, ionisches-Fe, Laktoferrin), gleich ist.
Regulation der Eisenabsorption
Beim Menschen ist sowohl die Häm- als auch die nicht-Häm-Eisenabsorption regulierbar, sodass im starken Eisenmangel
deutlich höhere Mengen Nahrungseisen absorbiert werden können. Der genaue Mechanismus ist sehr komplex, es gibt
verschiedene genetisch bedingte Erkrankungen, die direkt oder indirekt mit einer Fehlregulation der Eisenabsorption
einhergehen.
Eine Art der Regulation der Eisenabsorption muss über die Vorläuferzellen von Enterozyten in den Darmkrypten erfolgen,
die ausschließlich von der basolateralen Seite, also vom inneren Milieu des Körpers, mit Eisen versorgt werden und die
sich innerhalb von wenigen Tagen zu absorbierenden Zottenenterozyten verwandeln. Im Eisenmangel und bei hereditärer
Hämochromatose wird in den Kryptenzellen die DMT1-Aktivität über einen posttranslationalen Mechanismus gesteigert.
Dies geschieht über die Bindung eines "Iron-Responsive-Proteins (IRP) an ein "hairpin-IRE" in der 3'-nichttranslatierten
Region der mRNA von DMT1. Erhielt der Enterozyt in seiner Vorgeschichte ausreichend Eisen, wird die DMT1-Aktivität
herunterreguliert. Nach einer Hypothese zur Erklärung der physiologischen Funktion des HFE-Proteins, wurde diesem
eine Beteiligung an der Eisenversorgung der Kryptenenterozyten zugeschrieben (19).
Die offenbar effektivste Art der Regulation der intestinalen Eisenabsorption ist über das IREG1/Ferroportin möglich, das
für den Transport von Eisen aus Enterozyten ins Plasma zuständig ist (Abb.2). Nachdem C. Finch 1994 einen "storage-
iron regulator" postuliert hatte, wurde 2001 mit dem 25 AS-Peptid Hepcidin ein solcher Regulator des Eisenstoffwechsels
tatsächlich gefunden (20-23).
Literatur Eisenstoffwechsel
1.
WHO/NHD/01.3. Iron deficiency anemia assessment, prevention, and control. World Health Organization,
200
2.
Pietrangelo A. Hereditary hemochromatosis--a new look at an old disease. N Engl J Med
2004;350(23):2383-97
3.
Heilmeyer L, Plötner K. Eisenmangelzustände und ihre Behandlung. Klin Wochenschr 1936; 15:1669
4.
Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo RJr, Ellis MC,
Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland
E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Qiuntana L, Starnes SM, Schatzmann
RC, Brunke KJ, Drauna DT, Risch NJ, Bacon BR, Wolff RK. A novel MHC class I-like gene is mutated in
patients with hereditary haemochromatosis. Nature Genetics 1996; 13:399-408
5.
Bothwell TH, Charlton RW, Cook JD, Finch CA. Iron metabolism in man. Blackwell Scientific Publications,
1979
6.
Brock JH, Halliday JW, Pippard MJ, Powell LW. Iron metabolism in health and disease. WB Saunders
Company Ltd. London, Tokyo 1994
7.
McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D,
Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ. An iron-regulated ferric
reductase associated with the absorption of dietary iron. Science 2001; 291:1755–1759
8.
Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger
M. Cloning and charcterization of a mammalian proton-coupled metal-ion transporter. Nature 1997; 388:482-
488
9.
Garrick MD, Dolan KG, Horbinski C, Ghio AJ, Higgins D, Porubcin M, Moore EG, Hainsworth LN, Umbreit
JN, Conrad ME, Feng L, Lis A, Roth JA, Singleton S and Garrick LM. DMT1. A mammalian transporter for
multiple metals. BioMetals 2003; 16:41–54
10.
Hubert N, Hentze MW. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1.
implications for regulation and cellular function. Proc Natl Acad Sci U S A. 2002; 99:12345-50
11.
McKie AT, Martinai P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F,
Hediger MA, W. Hentze MW, Simpson RJ. A novel duodenal iron-regulated transporter, IREG1, implicated in
the basolateral transfer of iron to the circulation. Mol Cell 2000; 5:299-309
12.
Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law
TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI.
Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000;
403:776-781
13.
Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ. Hephaestin, a
ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet
1999; 21:195–199
14.
Conrad ME, and Umbreit JN. Iron absorption-The mucin mobilferrin integrin pathway. A competitive pathway
for iron absorption. Am J Hematol 1993; 42:67–73
15.
Hallberg L. Perspectives on nutritional iron deficiency. Annu Rev Nutr 2001; 21:1–21
16.
Worthington MT, Cohn SM, Miller SK, Luo RQ, Berg CL. Characterization of a human plasma membrane
heme transporter in intestinal and hepatocyte cell lines. Am J Physiol 2001; 280:G1172–G1177
17.
Weintraub LR, Conrad ME, Crosby WH. Absorption of hemoglobin iron by the rat. Proc Soc Exp Biol Med
1965; 120:840–843
18.
Boni RE, Huch Boni RA, Galbraith RA, Drummond GS, Kappas A. Tin-mesoporphyrin inhibits heme
oxygenase activity and heme-iron absorption in the intestine. Pharmacology 1993; 47:318–329
19.
Fleming RE, Sly WS. Mechanisms of iron accumulation in hereditary hemochromatosis. Annu Rev
Physiol.2002; 64:663-680
20.
Finch C. Regulators of iron balance in humans. Blood 1994; 84:1697-1702
21.
Krause A, Neitz S, Mägert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K. LEAP-1, a novel
highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett 2000; 480:147-150
22.
Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loreal O. A new mouse liver-specific gene,
encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron
overload. J Biol Chem 2001; 276:7811–7819.
23.
Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Lack of hepcidin gene
expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc
Natl Acad Sci USA 2001; 98:8780-8785











© www.eiseninfo.de
Eisenstoffwechsel
Eisen ist das vierthäufigste Element in der
gesamten Erde und in der kontinentalen
Erdkruste. Als Übergangsmetall hat Eisen
vielfältige chemische Reaktionsmöglichkeiten
und es ist sicher deswegen von der Evolution als
Spurenelement für fast alle bekannten Lebewesen
ausgewählt worden, obwohl zuviel an Eisen in
biologischen Systemen toxische Reaktionen
hervorrufen kann.
Um Eisen gefahrlos nutzen zu können wurde ein
kompliziertes System von feinregulierter
Aufnahme, Transport und
Speicherungsmechanismen geschaffen, damit im
Normalzustand eine ausgeglichene Eisenbilanz
sichergestellt wird.


Eisenmangel
altertümliche Therapie des Eisenmangels (wenig wirksam!)