

Abb. 1: Art und Häufigkeit von Symptomen bei Typ1 Hämochromatose zum zeitpunkt der Diagnose (Zahlen: eigene
Daten Nielsen 2000)
Bedingt durch die progressive Eiseneinlagerung in parenchymale Organe kann es meist im höheren Lebensalter (ab 40-
50 Jahren) zu vielfältigen klinischen Symptomen kommen (1). Die Leber-Eisenkonzentration ist ein guter Anhalt für das
Ausmaß der individuellen Eisenspeicherung. Es besteht eine Korrelation zwischen Leber-Eisen und der Häufigkeit von
Leberzirrhose, Diabetes und Hautpigmentierung. Dieser Zusammenhang ist nicht gegeben für die Arthropathie, die vor
allem die Metacarpophalangealgelenke der Finger betrifft und die in einigen Fällen erst nach erfolgter
Eisenentzugstherapie erstmals auftritt (2,3).
Häufigstes Symptom ist eine auffällige Müdigkeit und Erschöpfheit, die manchen Arzt auch auf den Verdacht eines
Eisenmangels bringen kann. Wird dann ein erhöhtes Ferritin gemessen ist der Weg zur richtigen Diagnose dann eher
leicht.
Bei ca. 30% der Patienten entwickelt sich eine Arthropathie (Gelenkerkrankung), die manchen Patienten zum Arzt führt.
Die kleinen Gelenke der Hand (vor allem Fingergeleneke im Zeige- und Ringfinger) sind häufig als erste betroffen.
Patienten mit individuell schwerer Eisenüberladung zeigen auch eine eigentümliche metallisch-graue Pigmentierung der
Haut, die deutlich anders ist als eine gesunde Hautbräunung. Wer viele Patienten gesehen hat, erkennt daran meist
sofort einen Hämochromatose-Patienten.
Auffällig ist, dass zum Zeitpunkt der Diagnosestellung die Häufigkeit von schweren und irreversiblen Schäden
(Leberzirrhose, Diabetes) bei Diagnosestellung in den letzten Jahren stark rückläufig ist. In unserem eigenen
Patientenkollektiv, das in den letzten Jahren diagnostiziert worden ist, findet sich nur noch in ca. 10 % der Fälle eine
Leberzirrhose. Dies ist zum einen auf eine verbesserte Diagnostik und frühzeitige Therapie zurückzuführen, zum anderen
scheint die Penetranz der HFE-assoziierten Hämochromatose eher gering zu sein, sodass man heute auch zunehmend
viele klinisch wenig betroffene Patienten findet. Viele genetisch betroffenen Patienten werden heute nur aufgrund
erhöhter Blutparameter und des dann folgenden Gentests identifiziert, klinische Symptome zeigen sie (noch) nicht.
Zwei große Studien wiesen eine vergleichsweise niedrige klinische Ausprägung bei in Screeningstudien neu entdeckten
homozygoten C282Y-Trägern nach (4,5). Tab. 2:
Klinische Symptome
Niederau et al. 1985(6)
Adams et al. 1997 (7)
Nielsen 2000 (8)
Leberzirrhose
69 %
22 %
10 %
Diabetes
14 %
5 %
Arthropathie
43 %
29 %
35 %
Impotenz
55 %
40 %
5 %
Hautkolorierung
75 %
38 %
22 %
asymptomatisch
27 %
35 %
keine Eisenüberladung
15 %
Diese neueren Erkenntnisse sollten insgesamt keinesfalls dazu führen, die Typ1 Hämochromatose insgesamt zu
verharmlosen. Einzelne Patienten sind bereits in frühen Jahren schwer betroffen und weisen substantielle eiseninduzierte
Organschäden auf, sodass in jedem diagnostizierten Fall eine konsequente, vorsorgliche Eisenentzugstherapie durch
Aderlässe erfolgen sollte.
Die C282Y-Homozygotie ist der einzig gängige Genotyp, der einen klinisch relevanten Hämochromatose-Phänotyp
produziert. Andere Mutations-Konstellationen (C282Y/H63D-Compound-Heterozygotie, H63D-Homozygotie), führen nur
in wenigen Einzelfällen zu signifikanten biochemischen oder klinischen Symptomen in Richtung Eisenüberladung. Seit
der Identifizierung des HFE-Gens 1996 hat es viele Untersuchungen zu möglichen modifizierende Faktoren des
phenotypischen Ausprägungsgrades der Hämochromatose gegeben. Dabei wurden äußere Einflussfaktoren (Ernährung,
Alkohol, Drogen, metabolisches Syndrom) genauso diskutiert wie genetische Ursachen (häufige Polymorphismen in der
BMP-Hepcidinaktivierungskaskade) (9). Aktuell werden auch Variationen in der Hepcidin-Promotorregion diskutiert (10),
die zu einem schweren Phänotyp mit besonders niedrigen Hepcidinspiegeln führen (11).
Literatur 1. Pietrangelo A. Hereditary Hemochromatosis - A new look at an old disease. New Engl J Med 2004; 350:2383-2397 2. Schumacher HR. Hemochromatosis and Arthritis. Arthritis Rheumatol 1964; 7:41-50 3. Rihl M, Kellner H. Die Arthropathie der Hereditären Hämochromatose. Z Rheumatol 2004; 63:22–29 4. Niederau C, Fischer R, Sonnenberg A, Stremmel W, Trampisch HJ, Strohmeyer G. Survival and causes of death in cirrhotic and non-cirrhotic patients with primary haemochromatosis. N Engl J Med 1985; 313:1256-1262 5. Adams PC, Deugnier Y, Moirand R, Brissot P. The relationship between iron overload, clinical symptoms, and age in 410 patients with genetic hemochromatosis. Hepatology 1997; 25:162-6 6. Nielsen P. Gendiagnostische Möglichkeiten der hereditären Hämochromatose. In: Handbuch der Molekularen Medizin, Band 6. Monogen bedingte Erbkrankheiten. D. Ganten, K. Ruckpaul (Hrsg.) Springer-Verlag Berlin Heidelberg 2000; 455-475 7. Beutler E, Felitti VJ, Koziol JA, Ho JN, Gelbart T. Penetrance of 845G*A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet 2002; 359:211-18 8. Asberg A, Hveem K, Thorstensen K, Ellekjter E, Kannelonning K, Fjosne U, Halvorsen TB, Smethurst HB, Sagen E, Bjerve KS: Screening for hemochromatosis: high prevalence and low morbidity in an unselected population of 65,238 persons. Scand J Gastroenterol 2001; 36:1108-1115 9. Deugnier Y, Mosser J. Modifying factors of the HFE hemochromatosis phenotype. Expert Rev Gastroenterol Hepatol. 2008 Aug;2(4):531-40. 10. Island ML, Jouanolle AM, Mosser A, Deugnier Y, David V, Brissot P, Loréal O. A new mutation in the hepcidin promoter impairs its BMP response and contributes to a severe phenotype in HFE related hemochromatosis. Haematologica. 2009 Mar 13. [Epub ahead of print] Links 11. Casanovas G, Mleczko-Sanecka K, Altamura S, Hentze MW, Muckenthaler MU. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J Mol Med 2009 May;87(5):471-80. doi: 10.1007/s00109-009-0447-2.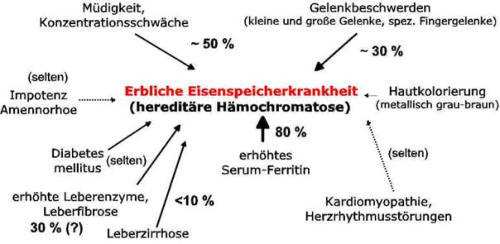












© www.eiseninfo.de
Symptome bei Eisenüberladung
Klinische Symptomatik
Im folgenden werden die Symptome bei der erblichen
Eisenspeicherkrankheit detailierter besprochen, die viele
Patienten zum Arzt führen und deshalb für die
Diagnosestellung wichtig sind. Bei transfusionsbedinger
Eisenüberladung ist die Diagnose meist bereits bekannt,
bevor es zum Teil zu den ähnlichen Symptomen
bezüglich Leber und Bauchspeicheldrüse kommt (s. sek.
Eisenüberladung, Thalassämie). Andere Symptome sind
sehr charakteristisch für die Hämochromatose, wie z.B.
die Gelenkbeschwerden (s. unten).

Eisenüberladung
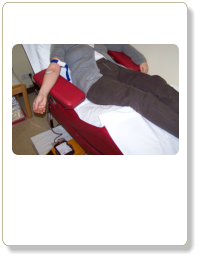
Aderlasstherapie. Methode aus dem Mittelalter!? Für Eisenspeicherkrankheit aber hochmodern