Typ1 HFE-Protein mutiert. Häufigste Form in Nordeuropa, USA, Australien (1 : 200-300); autosomal
rezessiv; parenchymale Eisenspeicherung: variabler, häufig milder Phänotyp (Leber, Gelenke, endokrine
Organe); spricht gut auf Aderlass an.
Typ 2A HJV-Protein mutiert
Typ 2B HAMP-Gen betroffen. Beides seltene Formen, Juvenile Hämochromatose, autosomal rezessiv; früh
einsetzende Eisenüberladung: häufig schwere Organschäden (Leber, endokrine Organe); spricht gut auf
Aderlass an.
Typ 3 TfR2-Protein betroffen. Sehr ähnlich Typ1. Häufiger in Südeuropa, autosomal rezessiv; parenchymale
Eisenspeicherung: variabler, häufig milder Phänotyp (Leber, Gelenke, endokrine Organe); spricht gut auf
Aderlass an.
Typ 4 (A und B) Ferroportin-Gen betroffen. Selten. Autosomal dominant; retikuloendotheliale
Eisenspeicherung: meist geringe Organbeteiligung, spricht teilweise schlecht auf Aderlass an.
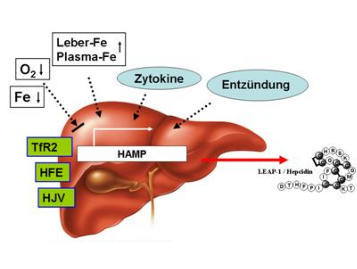
Abb. 1: Regulation der Hepcidinsynthese (HAMP-Gen) in der Leber. Mutationen im HFE-, TfR2- und HJV-Gen
(Typ 1-3 Hämochromatose) beeinträchtigen dabei direkt die Biosynthese von Hepcidin in der Leber.
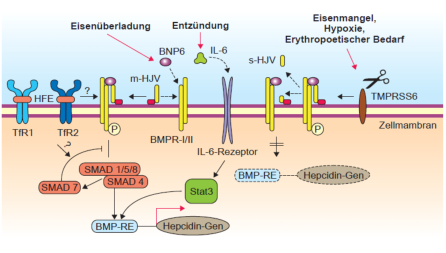
Abb. 2: Signaltransduktion der Hepcidinsynthese in der Leber. Das HFE-Protein, Transferrin-Rezeptor 2 und
Hämojulelin (HJV) sind daran beteiligt. Auch andere Stimuli wie Entzündung führen zu einer Hepcinsynthese.
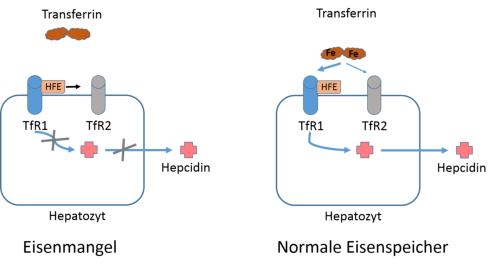
Abb. 3: Model der Hepcidinsynthese in Hepatozyten. Hepatozyten haben den Transferrinrezeptor 1 (TfR1) und 2 (TfR2)
exprimiert, die sich an der Obefläche befinden. TfR1 hat eine wesentlich höhere Affinität zu Transferrin als TfR2 und beide
können das HFE-Protein binden. Bei Eisenmangel bindet kein Transferrin an TfR1 und das HFE-Protein wird an TfR2
abgegeben. In dieser Situation wird physiologisch kein Hecidin gebildet und die intestinale Eisenabsorption bleibt
hochreguliert. Bei hohem Serum-Eisen bindet Transferrin vorallem an TfR1 und es wird im Hepatocyten Hepcidin gebildet.
Dieses Modell erklärt die Beteiligung von HFE und TfR2 an Formen der Hämochromatose (Typ1 bzw. Typ 3), die auf einer
Störung der Hepcidinsynthese beruhen. Model nach Montalbetti et al. 2013.
Juvenile Hämochromatose (Typ 2A und B)
Die juvenile Hämochromatose ist eine seltene Form einer Eisenüberladung, die sich durch einen frühzeitigen
Beginn (vor dem 30. Lebensjahr), durch ausgeprägte klinische Symptome (hypogonadotropher
Hypogonadismus, Kardiomyopathie, Leberzirrhose), sowie eines allgemein schweren Verlaufes unterscheidet.
Todesfälle durch Herzversagen sind häufig (27). Die meisten Fälle (Typ 2A) mit juveniler Hämochromatose
sind genetisch verbunden mit dem Lokus 1q21 (28) . Das Gen wurde inzwischen kloniert (LOC148738) (29).
Es kodiert für ein Protein, Hämojuvelin, dessen Funktion bisher nicht bekannt ist. Überraschenderweise gibt es
einige Patienten (ca. 10 %) mit den gleichen klinischen Symptomen, die keine Mutationen in diesem Genlokus
haben, sondern im HAMP-Gen (19q13.1; Typ 2B), das für Hepcidin kodiert (30). Damit liegt nahe, dass
Hämojuvelin direkt etwas mit Hepcidin, z.B. mit der Modulation der Hepcidin Expression zu tun haben muss.
Das einheitliche Erscheinungsbild der Erkrankung mag auf einer altersabhängigen Empfindlichkeit der Organe
für stark erhöhtes Eisen, z.B. in Form von NTBI beruhen oder auf einer besonderen Verteilung von
überschüssigem Eisen in speziellen Zellen (31). Grundsätzlich sprechen diese Patienten gut auf
Aderlasstherapie an (8). Langzeitstudien gibt es bei den wenigen bekannten Patienten nicht. Die
Herztransplantation könnte in sonst aussichtslosen Fällen eine Therapieoption darstellen (27).
Transferrin-Rezeptor2- assoziierte Hämochromatose (Typ 3)
Bisher sind nur wenige Fälle beschrieben, in denen Mutationen im TfR2 die Ursache für eine hereditäre
Hämochromatose darstellen (32). TfR2 hat eine unklare Funktion im Eisenstoffwechsel, es kann genau wie
TfR1 Diferric-Transferrin binden und internalisieren. Dies scheint aber nicht die physiologische Funktion zu
sein, denn Mutionen im TfR2 führen zur Eisenüberladung und nicht zur Eisendepletion der Leber. Zwei
alternative ( und ) Splice-Formen von TfR2 sind isoliert worden, das -Transkript wird hauptsächlich in der
Leber exprimiert. Auch bei dieser Form der Hämochromatose sind ganz geringe oder fehlende Hepcidin-
Konzentrationen im Urin zu messen (33).
Die Klinik der Typ3-Hämochromatose ist ähnlich dem HFE-assoziierten Verlauf mit erhöhten Ferritinwerten, in
Einzelfällen auch Leberzirrhose, Arthropathie. Diese Form spricht ebenfalls gut auf eine Aderlasstherapie an.
Ferroportin- assoziierte Hämochromatose (Typ 4)
Die Ferroportin-Krankheit (Hämochromatose Typ 4) ist eine neu entdeckte Form der erblichen
Eisenspeicherkrankheit, die charakterisiert ist durch Eisenspeicherung in Makrophagen inkl. Kupfferzellen in
der Leber (34). Ursache ist eine Mutation im Ferroportin-Gen (am häufigsten V162del) (34, 35). Die Patienten
haben Hämosiderinablagerungen in allen Geweben (mesenteriale Lymphknoten, Leber, Magen- und
Dünndarmmukosa). Ferritin ist frühzeitig erhöht (> 1000 µg/l). Anzeichen von Organschäden gibt es auch im
hohen Alter nicht. Es finden sich erhöhte Konzentrationen von Pro-Hepcidin im Blut, die auch mit Hepcidin im
Urin korrelieren. Hepcidin ist in diesen Fällen offenbar wirkungslos in der Hemmung der
Nahrungseisenabsorption bei gefüllten Eisenspeichern (36).
Neonatale Hämochromatose
Die neonatale Hämochromatose (NC) ist die häufigste Ursache für ein neonatales Leberversagen. Die
Häufigkeit wird mit 10-20 Fällen pro 100.000 Lebendgeburten/Jahr angegeben. Es besteht immer eine starke
Hyperferritinämie, fast alle Kinder weisen eine lebensbedrohliche schwere Lebersiderose auf. Trotzdem liegt
wohl keine genetisch bedingte Eisenüberladung vor. Nach Whitington et al. besteht eine Alloimmunität gegen
ein fetales Antigen (37). Es erkranken nur immer nur Neugeborene von bestimmten Müttern, nicht aber von
bestimmten Vätern. Bei weiteren Schwangerschaften besteht ein hohes Risiko (80 %) für weitere Fälle.
Allerdings steht mit der hochdosierten Immunglobulin-Therapie (1 g/kg wöchentlich ab der 18
Schwangerschaftswoche) offenbar eine wirksame Therapie zur Verfügung (37) Fälle mit NC werden in erster
Option mit einem antioxidativen Cocktail und Deferoxamin behandelt. Schlägt diese Therapie nicht an, kann
nur noch eine Lebertransplantation helfen (38).
Afrikanische Eisenüberladung
Die Afrikanische Eisenüberladung (African iron overload, AIO) wird in Ländern südlich der Sahara beoachtet
(2, 39). Sie ist charakterisiert durch die überschüssige Speicherung von Eisen vorwiegend in
reticuloendothelialen Zellen der Leber, Milz, Knochenmark aber auch in Hepatozyten. Die Histologie ist klar
unterschiedlich zu der HFE-assoziierten Hämochromatose. Früher wurde AIO unter dem Begriff Bantu-
Siderose allein auf das Trinken großer Mengen traditionell gebrauten Bieres zurückgeführt, das einen hohen
Milchsäuregehalt hat, was offenbar Eisen aus den Eisengefäßen herauslösen kann (40). Die
Eisenkonzentration lag bei zu 100 mg/Liter bei einem geringen Alkoholanteil (1-3 %) und es wurden
üblicherweise mehrere Liter/Tag getrunken. Neuere Arbeiten legen jedoch eine genetische Ursache nahe, die
zusammen mit der eisenreichen Nahrung zu AOI führt. Ein Defekt im Ferroportin könnte eine mögliche
Ursache sein. In einigen Gegenden sind bis zu 10 % der Bevölkerung betroffen (40).
Porphyria Cutanea Tarda
Porphyria cutanea tarda (PCT) ist die häufigste Form der Porphyrie (Prävalenz: 1 auf 5000-25000). PCT
entsteht, wenn die Uroporphyrinogen Decarboxylase, ein Leberenzym, das zur Hämsynthese notwendig ist,
inaktiv ist (41). PCT umfasst eine familiäre Form mit Mutationen im UROD-Gen sowie eine erworbene Form die
Individuen mit einer nicht näher charakterisierten genetischen Prädisposition umfasst. (sporadische PCT), die
nach Exposition mit Hepatotoxinen aktiv werden kann. Das Ergebnis ist in jedem Fall eine Akkumulation von
Porphyrinen in der Haut, die zu einer Photosensitivität und damit zu einer Schädigung der Haut führt. Es gibt
bekannte erworbene Faktoren wie Alkoholkonsum, Hepatitis C-Infektion, Östrogentherapie, die eine PCT
auslösen oder verschlimmern können. Zu diesen Faktoren gehört offenbar auch Eisenüberladung (41). In einer
Studie an 108 Patienten mit PCT waren 19 % homozygot für die C282Y-Mutation (42). Jede neu entdeckte
Patient mit PCT sollte deshalb auf HFE-assozierte Hämochromatose untersucht werden. Nach eigenen
Erfahrungen sind Patienten mit PCT im positiven Sinne sehr empfindlich auf eine Eisenentzugstherapie. Hier
sollte die Erhaltungstherapie sehr strikt durchgeführt werden (Ferritin stets < 30 µg/l).
Atransferrinämie
Die hereditäre Atransferrinämie ist eine extrem seltene Krankheit (weltweit weniger als 10 Fälle bekannt), die
offenbar durch verschiedene Mutationen im Transferrin-Gen (3q21) hervorgerufen wird (43). Sie ist
charakterisiert durch eine mikrozytäre Anämie im frühen Kindesalter und der gleichzeitigen Entwicklung einer
Eisenüberladung. Heilmeyer hat 1961 den ersten Fall beschrieben, ein 7-Jahre altes Mädchen mit schwerer
hypochromer Anämie, das an Herzversagen verstarb (44). Die Autopsie erbrachte eine schwere Siderose in
Herz und Leber. Nur Spuren von Transferrin im Plasma waren mit immunologischen Methoden nachweisbar,
beide Elternteile wiesen die Hälfte der normalen Transferrinkonzentration auf. Eine Behandlungsmöglichkeit ist
durch regelmäßige Plasmainfusionen gegeben. Als Tiermodel ist eine Hypotransferrinämie-Maus (trf[hpx]) mit
schwerer Anämie, hochregulierter Eisenabsorption und Lebersiderose bekannt (45).
Acaerulosplasminämie
Caeruloplasmin (Cp) ist eine Multi-Kupfer-Ferroxidase, die den größten Anteil vom Serum-Kupfer bindet (46).
Die eigentlich wichtigen Funktionen liegen aber nicht im Kupfer- sondern im Eisenstoffwechsel. CP hat eine
Funktion in der Freisetzung von Eisen aus Zellen und bei der Inkorporation von absorbiertem Eisen in Apo-
Transferrin (47). Die Acaeruloplasminämie ist eine seltene autosomal rezessiv vererbte Erkrankung, die durch
Mutationen im CP-Gen bedingt ist. Der Mangel von CP führt zur Eisenakkumulation in Neuronen und
Astrozyten im Gehirn, in der Retina, in parenchymalen und retikuloendothelialen Zellen der Leber, Milz und
Pankreas. Meist im Erwachsenenalter erkranken die Patienten an Diabetes, Ataxie und Demenz und retinaler
Degeneration. Eine Aderlasstherapie ist nicht sinnvoll und verschlimmert die ebenfalls bestehende Anämie.
Eine Therapie mit Deferoxamin ist beschrieben (48). Dadurch wird offenbar erhöhtes Lebereisen entfernt, nicht
aber erhöhte Eisenspeicher im Gehirn. Auch scheint die Anämie durch die Desferaltherapie verstärkt zu
werden.
Literatur27.
Kaltwasser JP, Gottschalk R, Seidl CH: Severe juvenile haemochromatosis (JH) missing HFE gene variants: implications
for a second gene locus leading to iron overload. Brit J Haematol 1998; 102:1111-1122
28.
Roetto A, Totaro A, Cazzola M, Cicilano M, Bosio S, D'Ascola G, Carella M, Zelante L, Kelly AL, Cox TM, Gasparini P,
Camaschella C. Juvenile hemochromatosis locus maps to chromosome 1q. Am J Hum Genet. 1999; 64:1388-1393
29.
Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, Andres L, MacFarlane J,
Sakellaropoulos N, Politou M, Nemeth E, Thompson J, Risler JK, Zaborowska C, Babakaiff R, Radomski CC, Pape TD, Davidas
O, Christakis J, Brissot P, Lockitch G, Ganz T, Hayden MR, Goldberg YP. Mutations in HFE2 cause iron overload in chromosome
1q-linked juvenile hemochromatosis. Nat Genet. 2004; 36:77-82
30.
Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, Loukopoulos D, Camaschella C. Mutant
antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003; 33: 21-22
31.
Lee PL, Beutler E, Rao SV, Barton JC. Genetic abnormalities and juvenile hemochromatosis: mutations of the HJV gene
encoding hemojuvelin. Blood 2004; 103:4669-4671
32.
Roetto A, Totaro A, Piperno A, Piga A, Longo F, Garozzo G, Cali A, De Gobbi M, Gasparini P, Camaschella C. New
mutations inactivating transferrin receptor 2 in hemochromatosis type 3. Blood 2001; 97:2555-2560
33.
Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C: Hepcidin is decreased in TFR2 hemochromatosis Blood
2005; 105:1803-1806
34.
Njajou OT, Vaessen N, Joosse M, Berghuis B, Dongen JWF van, Breuning MH, Snijders PJLM, Rutten WPF, Sandkuijl
LA, Oostra BA, Duijn CM van, Heutink P. A mutation in SLC11A3 gene is associated with autosomal dominant hemochromatosis.
Nature Genet 2001; 28:214-215
35.
Montosi G, Donovan A, Totaro A, Garuti C, Pignatti E, Cassanelli S, Trenor CC, Gasparini P, Andrews NC, Pietrangelo A.
Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest 2001;
108:619-623
36.
Zoller H, McFarlane I, Theurl I, Stadlmann S, Nemeth E, Oxley D, Ganz T, Halsall DJ, Cox TC, Vogel W. Primary Iron
Overload With Inappropriate Hepcidin Expression in V162del Ferroportin Disease. Hepatology 2005; 42:466-472
37.
Whitington PF, Kelly S, Ekong UD. Neonatal hemochromatosis: Fetal liver disease leading to liver failure in the fetus and
newborn. Pediatr Transplantation 2005; 9:640-645
38.
Rodrigues F, Kallas M, Nash R, Cheeseman P, D'Antiga L, Rela M, Heaton ND, Mieli-Vergani G. Neonatal
hemochromatosis--medical treatment vs. transplantation: the king's experience. Liver Transpl 2005; 11: 1417-24
39.
Moyo VM, Mandishona E, Hasstedt SJ, Gangaidzo IT, Gomo ZAR, Khumalo H, Saungweme T, Kiire CF, Paterson AC,
Bloom P, MacPhail AP, Rouault T, Gordeuk VR. Evidence of genetic transmission in African iron overload. Blood 1998; 91:1076-
1082
40.
Gordeuk VR African iron overload Semin Hematol. 2002; 39:263-269
41.
Sampietro M, Fiorelli G, Fargion S: Iron overload in porphyria cutanea tarda. Haematologica 1999; 84:248-253
42.
Chiaverini C, Halimi G, Ouzan D, Halfon P, Ortonne JP, Lacour JP: Porphyria cutanea tarda, C282Y, H63D and S65C
HFE gene mutations and hepatitis C infection: a study from southern France. Dermatology 2003; 206(3):212-6
43.
Beutler E, Gelbart T, Lee P, Trevino R, Fernandez MA, Fairbanks VF,. Molecular characterization of a case of
atransferrinemia. Blood 2000; 96: 4071-4074
44.
Heilmeyer L, Keller W, Vivell O, Keiderling W, Betke K, Wohler F, Schultze HE. Kongenitale Atransferrinaemie bei einem
sieben Jahre alten Kind. Dtsch Med Wschr 1961; 86: 1745-1751
45.
Craven, C. M.; Alexander, J.; Eldridge, M.; Kushner, J. P.; Bernstein, S.; Kaplan, J. :
Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: a rodent model for
hemochromatosis. Proc Nat Acad Sci 1987; 84:3457-3461
46.
Nittis T, Gitlin JD. The copper-iron connection: hereditary aceruloplasminemia. Semin Hematol 2002; 39:282–289
47.
Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular
iron efflux. Proc Natl Acad Sci USA 1999;96:10812–10817
48.
Mariani R, Arosio C, Pelucchi S, Grisoli M, Piga A, Trombini P, Piperno A: Iron chelation therapy in
aceruloplasminaemia: study of a patient with a novel missense mutation
Gut 2004; 53:756– 758











© www.eiseninfo.de
Hämochromatose Typ 2 - 4
Nach der Entdeckung des C282Y-Mutation im HFE-
Gen als Ursache der Typ1 Hämochromatose wurden
weitere genetische Formen der Hämochromatose
aufgeklärt, die alle etwas mit der Synthese oder der
Funktion von Hepcidin zu tun haben (Tabelle 1, Abb. 1-
3)

Eisenüberladung
Aderlasstherapie. Methode aus dem Mittelalter!? Für Eisenspeicherkrankheit aber hochmodern