Abb.1 Weltweite Verbreitung der Thalassämien (a-Form in Asien, ß-Form im Mittelmeerbereich). 
Der erste Name für diese Krankheit („Cooley’s Anemia“), die bei Kindern mit einer schweren Anämie in Verbindung mit
Splenomegalie sowie charakteristischen Knochenveränderungen einhergeht, trägt den Erstbeschreibern Rechnung (4). Da
anfangs alle Patienten nur aus Mittelmeerländern zu stammen schienen, bürgerte sich schnell der Name „Thalassämie“
nach dem griechischen Wort für Meer (alassa) ein. Die Thalassämien gehören ähnlich wie die Hämochromatose zu den
häufigsten monogenen Erbkrankheiten überhaupt. Etwa 1,67 Prozent der Weltbevölkerung sind Träger einer a- oder b-
thalassämischen Mutation (5). In Deutschland leben derzeit etwa 450-500 Patienten mit einer b-Thalassaemia major, 100
Patienten mit einer ß-Thalassaemia intermedia. Angesichts der derzeitigen Bevölkerungszusammensetzung ist davon
auszugehen, dass etwa 150.000 Mitbürger heterozygote Träger („Minor“-Form) einer Hämoglobinerkrankung sind. Bei den
in Deutschland lebenden Migranten sind -Thalassämien zwar weitaus seltener als -Thalassämien, gleichwohl ist die Zahl
der diagnostizierten Patienten stark steigend. Bislang sind insgesamt etwa 2000 Patienten mit einer -Thalassämie bekannt
geworden (in der Mehrzahl Minor-Formen), darunter 180 deutsch-stämmige Kinder bzw. Erwachsene.
Den a-Thalassämie-Syndromen liegt eine Störung der quantitativen Synthese von a-Globinketten zugrunde. Sie kommen
wie die ß-Thalassaemie in hoher Frequenz in den subtropischen Malaria-Endemiegebieten vor, überwiegend jedoch in der
Bevölkerung Asiens, Arabiens und Afrikas, weniger häufig im Mittelmeerraum. Molekulare Ursache ist meistens eine
partielle oder totale Deletion eines oder mehrerer der insgesamt vier -Globingene, welche die -Ketten-Produktion regulieren.
Im folgenden wird nur auf die in Europa vorherrschende Form der ß-Thalassämie eingegangen.
Die ß-Thalassämie
Genetische Grundlagen und klinische Einteilung
Der molekulare Defekt besteht in einer Vielzahl unterschiedlicher Mutationen innerhalb des ß-Globingens, die eine partielle
(ß+-Thalassämie) oder totale (ß0-Thalassämie) Inaktivierung eines der beiden ß-Globingene hervorrufen. Die Inaktivierung
der ß-Globingene erfolgt am häufigsten durch Punktmutationen, von denen bisher weit über 200 verschiedene Varianten
identifiziert wurden. Sie betreffen alle Schritte der Genexpression. Seltener sind Gendeletionen (3,6).
Nach klinischen Gesichtspunkten erfolgt die Einteilung in Thalassaemia minor = heterozygote -Thalassämie, Thalassaemia
major = i.d.R. homozygote oder gemischt-heterozygote -Thalassämie mit Transfusionsabhängigkeit und Thalassaemia
intermedia = meist homozygote oder gemischt-heterozygote -Thalassämie mit zusätzlichen genetischen Veränderungen,
die zu einer Abmilderung der für die Thalassaemia major typischen Klinik führen (2-7).
Thalassaemia major
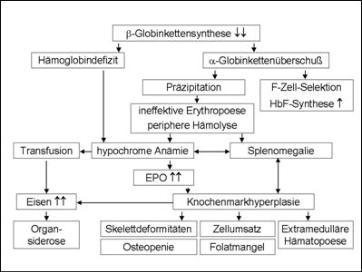
Die Pathophysiologie der Thalassaemia major basiert auf einer schweren Imbalance der Synthese von Polypeptidketten
(Abbildung 2). Neben dem Mangel an b-Ketten besteht ein Überschuss an a-Ketten, der nicht ausreichend durch die
Synthese von g-Ketten kompensiert werden kann und zu einer enorm gesteigerten Apoptose erythroider Vorläuferzellen
führt (4,8,9). Verminderte Hämoglobinsynthese, intravasale Hämolyse, Sequestration der Erythrozyten in der Milz und
ineffektive Erythropoese verursachen die Anämie. Die Folge ist eine stark (bis zu 30fach!) gesteigerte, aber ineffektive
medulläre und extramedulläre Erythropoese mit enormer Ausweitung der blutbildenden Markräume (5).
Abb.2:
Schematische Darstellung der pathophysiologischen Veränderungen bei der ß-Thalassaemia major (Erklärung
s. Text)
Die Thalassaemia major führt meist bereits im Verlauf des ersten Lebensjahres zu den klinischen Symptomen Blässe,
Ikterus, Gedeihstörung und Hepatosplenomegalie (7). Bei unzureichender Therapie kommt es zu Wachstumsretardierung,
häufigen Infektionen und Knochendeformierungen, die u.a. zu einer charakteristischen sog. Facies thalassaemica (hohe
Stirn, Verbreiterung der Diploe, Prominenz von Jochbein und Oberkiefer) führen. Unter den hämatologischen Symptomen
dominiert die sehr schwere, mikrozytär-hypochrome und geringgradig hämolytische Anämie mit hochgradig ineffektiver
Erythropoese. Die Patienten sind lebenslang transfusionsbedürftig, unbehandelt sterben sie in der frühen Kindheit. Im Laufe
der Erkrankung entwickelt sich aufgrund der parenteralen Eisenzufuhr bei regelmäßiger Transfusionstherapie sowie der
wegen der gesteigerten Erythropoese erhöhten intestinalen Eisenresorption eine ausgeprägte Eisenüberladung
(Hämosiderose). Zur Verhinderung siderose-bedingter Organschäden ist eine Eiseneliminationstherapie erforderlich (s. u.).
Thalassaemia intermedia
Die Thalassaemia intermedia ist eine klinische Diagnose für eine Gruppe mittelschwerer Krankheitsbilder, die ohne
chronische Transfusionsbedürftigkeit verläuft. Die genetische Basis bilden homozygote oder gemischt-heterozygote b-
Thalassämien, deren phänotypische Manifestation durch verschiedene Einflussfaktoren modifiziert wird (homozygote oder
gemischt-heterozygote b-Globingen-Mutationen mit hohen Restaktivitäten, die Kombination einer Thalassaemia major mit
einer hereditären HbF-Persistenz (HPFH) oder thalassämischen a-Globingendeletionen, und dominante b-Thalassämien).
Die Diagnose wird meist nach dem zweiten Lebensjahr gestellt, wobei in der Regel ein Hämoglobinwert von über 8 g/dl
aufrechterhalten werden kann und Wachstum und Entwicklung zumindest initial ungestört verlaufen.
Weitere klinische Besonderheiten der Thalassaemia intermedia sind ein infolge der exzessiven Erythropoese entstehender
Folsäuremangel ( Substitutionstherapie), die Entstehung von Gallensteinen infolge der Hämolyse ( Cholezystektomie), bei
erwachsenen Patienten die Entwicklung von Unterschenkel-Ulzera sowie eine erhöhte Thromboseneigung, die u.a. auf eine
prokoagulatorische Aktivität der geschädigten Erythrozyten zurückgeführt werden. Darüber hinaus kann es zur Entstehung
von Pseudo-Tumoren extramedullärer Blutbildung kommen, die bevorzugt paravertebral gelegen sind und zu bedrohlichen
neurologischen Komplikationen führen können (10,11).
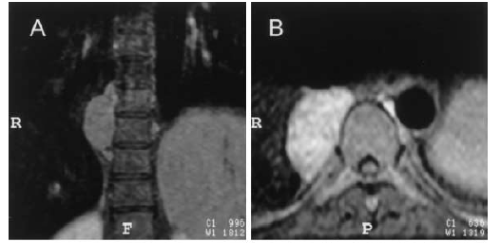
Abb. 3: T1-gewichtete coronare MR-Darstellung nach Gadolinium-Injektion (A) und transverse Fett-gesättige Darstellung
nach Gadolinium-Injektion (B). Darstellung einer solitären rechts paravertebral, scharf abgegrenzt gelegenen
Raumforderung in Höhe BWK 9/10, die sich aufgrund der extramedullären Blutbildung nach Gadolinium-Gabe iso- bis
hyperintens präsentiert (11).
Bei Auftreten von Komplikationen infolge der stark gesteigerten, jedoch ineffektiven Erythropoese ist im Einzelfall eine
Indikation zur Transfusionstherapie gegeben, wobei die klinische Situation und nicht der gemessene Hämoglobinwert
entscheidend ist (12). Auch Patienten ohne Transfusionstherapie entwickeln aufgrund der gesteigerten intestinalen
Eisenresorption im Krankheitsverlauf eine zumindest intermittierend therapiepflichtige Hämosiderose.
Thalassaemia minor
Patienten mit einer Thalassaemia minor haben in der Regel bei nur geringgradig erniedrigtem oder normalen
Hämoglobingehalt keine klinischen Symptome. Ein zusätzlicher Eisenmangel (bei etwa 10% der Kinder heterozygoter b-
Thalassämie, die wegen einer Anämie bzw. hypochromen Anämie untersucht werden) kann zu einer ausgeprägten Anämie
führen und muss dann wie bei sonstigen Eisenmangelanämien behandelt werden.
Behandlung der Thalassaemia major
Eine kurative Therapiemöglichkeit besteht in der hämatopoetischen Stammzelltransplantation. Bei HLA-identischen
Geschwistern gilt diese bei Patienten mit Thalassaemia major als Therapie der Wahl. Die in den letzten Jahren publizierten
Daten für eine Stammzelltransplantation von einem nicht-verwandten, HLA-identischen Fremdspender geben Anlass zu der
Hoffnung, dass diese für Patienten ohne HLA-identische Geschwister künftig zu einer gleichwertigen Therapieoption werden
kann (4, 13).
Die symptomatische Behandlung der Thalassaemia major beinhaltet eine regelmäßige Transfusionstherapie in Kombination
mit einer Chelattherapie zur Verhinderung einer bedrohlichen Eisenüberladung des Organismus.
Siderosebedingte Organschäden bei Thalassaemia major
Ohne effektive Chelattherapie ist eine progressive Hämosiderose unausweichliche Folge der Erkrankung, verursacht durch
die hohe intestinale Eisenresorption und die Eisenzufuhr mit den Transfusionen. Durch die Transfusionstherapie gelingt es,
die Anämie zu korrigieren und damit die Masse des hämatopoetischen Markes zu reduzieren, sodass die exzessiv
stimulierte, aber frustrane Eisenabsorption wieder herunterreguliert wird. Mit jeder Einheit transfundierten
Erythrozytenkonzentrates werden dem Körper etwa 200 mg Eisen zugeführt, die durchschnittliche tägliche Eisenzufuhr
eines regelmäßig transfundierten Thalassämiepatienten beträgt damit etwa 0,4-0,6 mg/kg. Bei einem 12-jährigen Patienten
unter regelmäßiger Transfusionstherapie kommt es so zur Akkumulation von mehr als 55 g Eisen in Geweben, die
normalerweise insgesamt ca. 2 g Eisen enthalten (14). Unter den Bedingungen einer regulär geführten
Transfusionstherapie ist mit einer Organschädigung ab einer zugeführten Menge von 500 g Erythrozyten/kg - das entspricht
etwa 500mg Eisen/kg - zu rechnen (15). Durch eine Chelattherapie wird das Maß der Eisenüberladung deutlich reduziert
und meist konstant gehalten. Trotzdem bleibt auch bei optimaler Therapie eine gewisse Restsiderose erhalten, die in
Abhängigkeit von ihrem Ausmaß zu den typischen eisenbedingten Organschäden (s. Abb. 4) führen kann.
Myokardsiderose
Herzerkrankungen stellen in der medizinischen Betreuung von Patienten mit Thalassaemia major nach wie vor die größten
Probleme dar, sie stellen auch die häufigste Todesursache. Mehr als zwei Drittel der Todesfälle einer Kohortenstudie aus
sieben italienischen Zentren waren entweder auf Herzversagen (60,8%) oder eine Arrhythmie (6,8%) zurückzuführen. In
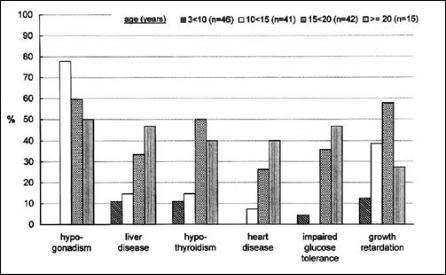
einer deutschen Studie waren 40% der Patienten im Alter von mehr als 21 Jahren von einer Herzerkrankung betroffen (Abb.
4) (16). Eine Ursache für Herzerkrankungen sind Probleme im Zusammenhang mit der Therapiecompliance, jedoch treten
leider auch bei Patienten mit scheinbar guter Therapieeinstellung, z.B. unter DFO-Therapie in bis zu 7 % kardiale
Erkrankungen auf (17).
Abb. 4:
Häufigkeit siderose-bedingter Organerkrankungen bei in Deutschland lebenden Patienten mit Thalassaemia
major (aus 11)
Arrhythmien und Erregungsleitungsstörungen treten häufig als erstes Symptom bei siderosebedingter Herzschädigung auf.
Neben isolierten ventrikulären und supraventrikulären Extrasystolen werden auch komplexere Rhythmusstörungen wie AV-
Blöcke I.-II. Grades, supraventrikuläre und ventrikuläre Tachykardien sowie Vorhofflattern beobachtet. Eine
antiarrhythmische Therapie ist in lebensbedrohlichen Verläufen notwendig, kann aber in einigen Fällen eine
Verschlimmerung der Arrhythmie zur Folge haben (18).
Der Herzinsuffizienz liegt eine Kardiomyopathie zugrunde, die sich als Myokardhypertrophie oder Ventrikeldilatation
manifestiert. Dabei geht die Störung der diastolischen Ventrikelfunktion meist der der systolischen voraus. Während die
Ruhefunktion des Herzens sehr lange erhalten zu bleiben scheint, lässt sich unter körperlicher oder pharmakologischer
Belastung (z.B. Ejektionsfraktion im Kardio-MRT) oft schon im Frühstadium eine Störung der Myokardfunktion erkennen.
Die Echokardiographie weist oft erst bei Auftreten erster Symptome der Herzinsuffizienz pathologische Befunde auf.
In den letzten Jahren haben sich durch Einführung neuer Diagnostikverfahren (T2*-MRT ) zunehmend Hinweise darauf
ergeben, dass bei einigen Patienten eine deutliche Dissoziation zwischen kardialer und hepatischer Eisenüberladung
besteht (19,20). Da in der Vergangenheit die Steuerung der Chelattherapie entscheidend von der Lebereisenkonzentration
abhing, könnte diese Dissoziation erklären, warum auch scheinbar gut chelierte Patienten an teilweise fatalen
Herzerkrankungen litten. Die T2*-MRT-Untersuchung weist auch prospektiv eine gute Korrelation mit kardialen
Funktionsparametern auf. Eine Stratifizierung bzgl. des Risikos, eine Herzinsuffizienz zu entwickeln erscheint anhand
definierter T2*-MRT-Werte möglich (21). In ersten auch randomisierten Studien mit verschiedenen Chelatbildnern erscheint
diese Methode als geeignet, zumindest über einen Zeitraum von mehreren Monaten bis einigen Jahren, die Effektivität
dieser Chelatbildner bzgl. der Entfernung myokardialen Eisens zu beurteilen (20,22).
Bei der Behandlung der Herzinsuffizienz bei Thalassämiepatienten sollten einige Besonderheiten beachtet werden. Eine
wichtige Rolle in der Therapie spielen ACE-Hemmer, die unter Beachtung der Blutdruckwerte dosiert werden. Für die
Behandlung mit Digoxin gilt, dass andere Erkrankungen, wie Hypothyreose oder Hypoparathyreodimus, ebenso wie die
Gefahr der Arrhythmien bei Thalassämiepatienten beachtet werden müssen. Eine suffiziente Überwachung der optimalen
Serumspiegel sowie der Serumelektrolyte ist notwendig. Bei der Gabe von Diuretika sollten vor allem Kalium-sparende
Diuretika zur Anwendung kommen (18).
Neben der symptomatischen Therapie ist vor allem in der Frühphase der Entstehung einer siderosebedingten
Kardiomyopathie die Intensivierung der Chelattherapie durch die Wahl eines anderen Chelators, eines anderen
Applikationsmodus, der Wahl einer höheren Dosis oder einer Kombination von verschiedenen Chelatoren von Bedeutung.
Eine besondere Problematik ergibt sich bei Patienten mit einer Myokardsiderose, die an einer Myokarditis erkranken. In
einer Studie mit 47 an einer Myokarditis erkrankten Thalassämiepatienten verstarben 22 Patienten an akutem
Herzversagen, ventrikulärer Tachykardie oder chronischem Herzversagen innerhalb der ersten Jahre nach Manifestation
der Myokarditis (23). Insbesondere für diese Patientengruppe mit schwerer lebensbedrohlicher Herzinsuffizienz stellt sich
die Frage nach der Indikation für eine Herztransplantation und deren Durchführbarkeit. Bislang liegen nur wenige
Einzelberichte vor, die allerdings die prinzipielle Möglichkeit belegen (24, 25). Sicher ist sowohl hinsichtlich der
Durchführbarkeit als auch bzgl. der Prognose die Frage weiterer Sideroseschäden (v.a. der Leber) von besonderer
Bedeutung.
Hepatopathie
Siderosebedingte Lebererkrankungen werden bei Thalassämiepatienten häufig (20 – 50 % der Patienten) dokumentiert,
wobei die Definition von Lebererkrankung unterschiedlich erfolgt. (16, 26, 27). Eine Zirrhose wird je nach untersuchtem
Patientenkollektiv in bis zu 10 % der Patienten diagnostiziert (26, 27). Vor allem in früheren Jahren stellte die Leberzirrhose
eine relevante Todesursache für Patienten mit einer b- Thalassämie dar (28). Die Bedeutung der siderosebedingten
Hepatopathie ergibt sich auch aus der Rolle der Leber z.B. in der IGF-1-Synthese ( Wachstummsstörung), bei Entstehung
eines Diabetes mellitus infolge einer erhöhten Insulinresistenz oder im Zusammenhang mit einem erhöhten
Thrombophilierisiko z.B. infolge Protein –C- oder –S-Mangel. Außerdem stellt das Ausmaß der Leberschädigung
(Hepatomegalie, Fibrose) einen prognostisch sehr wichtigen Parameter für die Prognosebeurteilung bei einer allogenen
Stammzelltransplantation dar (29).
Die Leberfibrose ist in ihrer Ausprägung sehr gut mit dem Lebereisengehalt korreliert und vor allem in frühen Stadien durch
eine adäquate Chelattherapie reversibel (30). Das Risiko der Entwicklung einer Leberfibrose steigt erheblich, wenn
zusätzlich eine chronische Hepatitis C vorliegt. So beträgt die Wahrscheinlichkeit des 10-Jahre-Fibrose-freien Überlebens
bei Patienten mit erheblicher Eisenüberladung und HCV-Negativität 40-50 %, dagegen weisen ca. 80 % der anti-HCV-
positiven Patienten nach 10 Jahren eine Leberfibrose auf (31). Die Prävalenz der HCV-Seropositivität bei Patienten mit
Thalassaemia major liegt in Abhängigkeit von dem untersuchten Patientenkollektiv und von der regionalen Herkunft
zwischen unter 20 % (Deutschland 22 %) bis hin zu 90 % (16, 26, 27).
Endokrine Störungen
Wachstumsstörungen
Unter adäquater Transfusions- und Chelattherapie ist das präpubertäre Wachstum von Patienten mit Thalassämie etwa bis
zum 10. Lebensjahr normal. Danach kommt es bei vielen Patienten zu einer Wachstumsverzögerung, insgesamt ist etwa
ein Drittel der Patienten von einer Wachstumsstörung betroffen (16, 32). Die verzögerte oder ausbleibende Pubertät trägt zu
der Wachstumsverzögerung entscheidend bei. Bei vielen Patienten kann ein partieller Wachstumshormonmangel
festgestellt werden, auch niedrige IGF-1-Werte, offenbar infolge mangelnder Lebersyntheseleistung, sind teilweise zu
finden. Neben Siderose-assoziierten Ursachen können auch Skelettschäden infolge der Chelattherapie zu einem
dysproportionierten Minderwuchs beitragen (32-34).
Neben der guten Dokumentation des Wachstums während der gesamten Kindheit und Adoleszenz sollten ab dem 10.
Lebensjahr jährliche IGF-1 und IGFBP3-Bestimmung sowie eine Untersuchung des Knochenalters erfolgen. Bei
Wachstumsverzögerung (Größe < 3.Pz.) sind diese Untersuchungen entsprechend früher indiziert. Bei auffälligen Befunden
schließen sich STH – Stimulationstests (Argininstimulation, Insulinhypoglykämie) an. Wichtig sind darüber hinaus die
Überprüfung der Dosis bei einer Chelattherapie mit Deferoxamin („therapeutischer Index“) sowie die Untersuchung
bezüglich anderer, potentiell zur Wachstumstörung beitragender Störungen (Hypothyreose, Hypogonadismus,
Glukoseintoleranz, Hepatopathie, Zöliakie).
Einige Patienten können möglicherweise von einer Wachstumshormontherapie profitieren. Es handelt sich hierbei jedoch
primär um einen experimentellen Therapieansatz. Vor Beginn einer solchen Therapie sollte der Anstieg von IGF-1 nach
Wachstumhormongabe nachgewiesen werden, um eine eventuell unwirksame Therapie zu vermeiden. Die Dosis orientiert
sich an den Richtlinien für die Therapie bei Wachstumshormonmangel anderer Ursache (12 IE/m2 KOF je Woche,
Dosiserhöhung während der Pubertät) (32,35).
Die rechtzeitige Induktion der ausbleibenden Pubertät durch Hormonsubstitutionstherapie kann zu einer
Wachstumsbeschleunigung beitragen (34). Allerdings besteht wegen der Gefahr des raschen Epiphysenschlusses hierbei
die Notwendigkeit der genauen Überprüfung der Indikationsstellung.
Hypogonadismus
Der Hypogonadismus stellt die häufigste endokrine Störung bei Patienten mit Thalassämie dar. Etwa die Hälfte der
Patienten (30-60 %) sind davon betroffen (16, 26, 28, 32). Das Spektrum reicht vom verzögerten Pubertätsbeginn über eine
ausbleibende bzw. unvollständige Pubertät bis zur sekundären Amenorrhoe bzw. sekundären Hypo- / Azoospermie (Tabelle
3.2). Ursache ist meist ein Ausfall der Hypothalamus-Hypophysen-Gonadenachse infolge einer Siderose des
Hypophysenvorderlappens, da dieser gegenüber siderosebedingt Radikal-induzierten Schädigungen sehr empfindlich ist
(36). Aber auch die siderotische Schädigung der Gonaden selbst kann ursächlich zu der Entwicklung eines
Hypogonadismus beitragen (36). Verschiedene Untersuchungen verweisen auf einen Zusammenhang zwischen
rechtzeitigem Beginn sowie zuverlässiger Durchführung der Chelattherapie und Ausmaß des Hypogonadismus (32,36,37).
Allerdings können auch einzelne Patienten mit scheinbar guter Chelattherapie einen Hypogonadismus entwickeln (38).
Zur frühzeitigen Diagnostik sind die jährliche Bestimmung der Gonadotropine, von Estradiol, Prolaktin, Testosteron und
SHBG für alle Patienten ab dem 10. Lebensjahr, sowie bei klinischen oder laborchemischen Auffälligkeiten ein LH-RH-Test,
bei Jungen ggf. gefolgt von einem HCG-Stimulationstest zu empfehlen. Für die Entscheidung zur Behandlung ist die
Beurteilung des Wachstums und des Knochenalters notwendig. Eine Hypothyreose muß ausgeschlossen bzw. behandelt
werden. Vor einem Knochenalter von 11 (Mädchen) bzw. 13 Jahren (Jungen) sollte noch nicht mit einer
Substitutionstherapie begonnen werden (32).
Bei positivem HCG-Stimulationstest kann bei männlichen Patienten eine subkutane Therapie mit HCG (z.B. Predalon®)
durchgeführt werden. Die Dosissteuerung erfolgt anhand der Serumtestosteronwerte sowie des klinischen Erfolges. Der
Vorteil dieser vom Patienten selbst durchführbaren Therapie besteht in der Stimulierung des Hodenwachstums durch HCG.
Bei Fertilitätswunsch ist die zusätzliche Gabe von HMG i.m. notwendig Bei negativem HCG-Stimulationstest erfolgt eine
Substitution von Testosteron in Form einer intramuskulären Injektionstherapie oder einer transdermalen Applikation (32,
36).
Bei Mädchen mit ausbleibender Pubertät wird in der Regel mit einer Therapie mit Östradiol in steigender Dosis begonnen,
später erfolgt der Zusatz eines Gestagens. Die Geschwindigkeit der Dosissteigerung sollte in Abhängigkeit vom Alter der
Patientin variiert werden. Auf das erhöhte Thromboserisiko ist besondere Aufmerksamkeit zu richten, vor allem in
Anbetracht möglicher Gerinnungsstörungen im Zusammenhang mit der Hämosiderose. Dies gilt insbesondere für
Patientinnen, die mit einem zentralvenösen Katheter versorgt sind.
Bei Jungen und Mädchen sollte etwa 6 Monate nach Beginn einer Pubertäts-induzierenden Therapie eine Reevaluation des
Hormonstatus erfolgen, bevor die Therapie bei entsprechender Notwendigkeit fortgesetzt wird.
Gestörte Glukosetoleranz und Diabetes mellitus
Ein sekundärer Diabetes mellitus entsteht bei etwa 5-10% der Patienten, zumeist manifestiert er sich in der zweiten
Lebensdekade (16, 26, 28). Der Manifestation des Diabetes geht in der Regel eine längere Phase mit erhöhter
Insulinresistenz und reaktiv vermehrter Insulinsekretion voraus (39, 40). Zusätzlich zu der dadurch bedingten sekundären
Pankreasinsuffizienz trägt als akzelerierender Faktor eine toxische Schädigung der pankreatischen Inselzellen durch die
Hämosiderose zur Entwicklung des Diabetes mellitus bei. Es besteht eine klare Beziehung zwischen der Störung der
Glukosetoleranz und Parametern der Eisenüberladung (Serumferritin, Lebereisen, Transfusionsmenge). Bei Patienten mit
einer schlecht durchgeführten Chelattherapie sind Störungen der Glukosetoleranz durch eine Intensivierung der
Chelattherapie häufig zumindest teilweise reversibel. Daher muß bei pathologischen Befunden initial immer die Effektivität
der Chelattherapie überprüft werden (32).
Jährliche Kontrollen des Nüchternblutzuckers, nach dem 10.Lebensjahr ergänzt durch jährlich durchzuführende orale
Glukostoleranztests, sind zu empfehlen. Bei der initialen Diagnostik aber auch beim eventuellen Therapiemonitoring ist zu
beachten, dass die HbA1C-Werte durch die Transfusionstherapie beeinflusst werden.
Bei noch erheblicher Restsekretion des Pankreas erfolgt die Therapie des Diabetes bei Thalassämie zunächst diätetisch,
ggf. unterstützt durch Maßnahmen zur Gewichtsreduktion, bei entsprechender Notwendigkeit durch zusätzliche Gabe oraler
Antidiabetika (Sulfonylharnstoffderivate, Acarbose). Sind mit dieser Therapie keine akzeptablen Blutzuckerwerte zu
erreichen, ist eine Insulintherapie zu beginnen. Eine frühzeitige Therapie mit Acarbose bei Patienten mit Hyperinsulinismus
aber noch normaler Glukosetoleranz kann den weiteren Verlauf möglicherweise positiv beeinflussen (41).
Hypothyreose
Die Entwicklung einer Hypothyreose unterschiedlichen Ausmaßes wird bei bis zu 25 % der Patienten mit Thalassaemia
major beobachtet. Die Angaben zur Häufigkeit schwanken in Abhängigkeit vom Ausmaß der Dysfunktion, dem Alter der
untersuchten Patienten sowie der verwendeten Therapieprotokolle (13, 16, 26, 28). Berücksichtigt man alle Schweregrade,
so weist etwa je ein Drittel der Patienten eine subklinische (pathologischer TRH-Test), eine kompensierte (erhöhtes TSH,
normales Thyroxin) oder eine dekompensierte Hypothyreose auf.
Es handelt sich in der Regel um eine primäre Hypothyreose bei Siderose der Schilddrüse, eventuell besteht eine
zusätzliche hypophysäre Komponente (Hypersensitivität der Hypothalamus-Hypophysen-Achse). Es besteht keine klare
Korrelation mit dem Serumferritin oder der Transfusionsmenge, wohl aber mit der Erhöhung der Lebertransaminasen.
Behandlungsbedürftig i.S.e. Substitutionstherapie sind sowohl die kompensierte als auch die dekompensierte
Hypothyreose. Bei erhöhtem T3, niedrigem T4 und erhöhtem TSH ist eine Jodmangelstruma auszuschließen.
Hypoparathyreoidismus
Die Häufigkeit des Hypoparathyreoidismus bei Patienten mit Thalassämie wird mit etwa 2-8% angegeben, wobei in den
letzten Jahren eine abnehmende Inzidenz zu beobachten ist (12, 16, 26). Symptome sind selten, können aber in einzelnen
Fällen lebensbedrohlich sein (42). Häufig liegen gleichzeitig Sideroseschäden anderer Organe vor. Die Therapie erfolgt mit
Calcitriol.
Osteopenie / Osteoporose / Osteopathie
Mit der zunehmenden Lebenserwartung von Patienten mit Thalassaemia major sind in den letzten Jahren neue Probleme in
den Mittelpunkt der Aufmerksamkeit getreten, die die Morbidität der Patienten entscheidend beeinflussen. Dazu gehören die
Knochenerkrankungen. Das Spektrum reicht von nur bei gezielter Untersuchung feststellbaren Veränderungen über
pathologische Frakturen bis hin zu schweren schmerzhaften Erkrankungen. In einer Untersuchung an 82 Patienten im Alter
von 12-43 Jahren wiesen 42 eine schwergradige, weitere 37 Patienten eine gering- bis mäßiggradige Reduktion der
Knochenmasse auf (43).
Eine Reihe von pathogenetischen Faktoren trägt zur Entstehung der Knochenerkrankungen bei. Dazu gehören die Anämie
und die damit verbundene Knochenmarkexpansion bei unregelmäßig transfundierten Patienten (vor allem bei Thalassaemia
intermedia), genetische Faktoren wie z.B. Polymorphismen im COLIA1-Gen (44), vor allem aber der oben beschriebene
Hypogonadismus. Andere Faktoren sind eine Eisenüberladung im Knochen, aber auch mögliche Schädigungen durch
Deferoxamin (chondrodystrophische Läsionen), IGF-1-Synthesestörungen, Hypoparathyreoidismus, Diabetes mellitus und
Hypothyreose ( 45, 46).
Wegen der langfristigen Bedeutung dieser Erkrankungen und wegen der Erfahrung, dass eine frühzeitige Behandlung für
den Behandlungserfolg insgesamt bedeutsam ist, sollten Patienten ab dem 10. Lebensjahr vor allem bei Präsenz eines
Hypogonadismus jährlich einer Knochendichtemessung (DEXA) unterzogen werden.
Die Behandlung der Osteopenie erfolgt durch Kalzium- und Vitamin-D-Supplementation. Bei Osteoporose ist der Einsatz
von Biphosphonaten zu erwägen, bisherige Studien zeigen einen deutlich Rückgang der Veränderungen, sowohl die
Knochendichte als auch Parameter des Knochenstoffwechsels betreffend (45, 46). Vor allem bei unregelmäßig
transfundierten Patienten (Thalassaemia intermedia) ist das Transfusionsschema zu überdenken oder aber eine
experimentelle Therapie mit Hydroxyharnstoff zu erwägen (47).
Betreuung von Patientinnen mit Thalassaemia major in der Schwangerschaft
Eine Schwangerschaft ist bei Thalassämiepatientinnen nach wie vor eine Seltenheit, systematische Daten hierzu sind nicht
verfügbar. Beispielhaft seien die Daten von zwei Übersichtsarbeiten erwähnt. Eine Publikation von Aessopos et al. aus dem
Jahre 1999 berichtete über 22 spontan (nicht durch Induktionstherapie) entstandene Schwangerschaften (1x Gemini) bei 19
Frauen mit b-Thalassämie, die 21 gesunde Kinder zur Welt brachten (48). Jensen et al. erfassten 1995 13
Schwangerschaften (1x Gemini) bei 11 Frauen, 9 davon spontan, 4 nach einer Induktionsbehandlung, 14 gesunde Kinder
wurden geboren (49).
Die Transfusionstherapie wird während der Schwangerschaft in der Regel in gleichen Abständen bei allerdings steigendem
Transfusionsbedarf fortgeführt. In der Richtlinie der Thalassemia International Federation wird empfohlen, den Hb-Gehalt
nicht unter 10 g/dl sinken zu lassen.
Keiner der bekannten Chelatbildner ist für die Behandlung in der Schwangerschaft zugelassen. Nach Bekanntwerden der
Schwangerschaft sollte jegliche Chelattherapie wegen der potentiellen Teratogenität der Medikamente zunächst
unterbrochen werden. Patientinnen, die mit Deferipron behandelt werden, wird empfohlen, bei Kinderwunsch auf
Deferoxamin zu wechseln. Überwiegend wird von erfahrenen Hämatologen die Meinung vertreten, dass eine Deferoxamin-
Behandlung (20-30 mg/kg/d) ab dem zweiten Trimenon möglich ist, es gibt zumindest keine Berichte über dadurch
verursachte Schäden bei Patientinnen, die so behandelt wurden bzw. ihren Kindern. Andere Kollegen verzichten während
der Schwangerschaft auf die Chelattherapie und heben hervor, dass die Siderose in dieser Zeit eindrucksvollerweise
weniger stark zunimmt als bei Patientinnen ohne Chelattherapie außerhalb der Schwangerschaft. Es gibt keinerlei
kontrollierte Daten zu diesem Thema. In der Stillzeit ist eine Therapie mit Deferoxamin (nicht mit Deferipron oder
Deferasirox) möglich, da davon auszugehen ist, dass Deferoxamin nicht über die Muttermilch übertragen wird.
Von besonderer Bedeutung bei der Betreuung schwangerer Thalassämiepatientinnen ist die Berücksichtigung sideroser-
bedingter Organschänden, vor allem der Herzinsuffizienz und der Glukosetoleranzstörung. Engmaschige
echokardiographische und endokrinologische (OGTT) Kontrollen vor allem im zweiten und dritten Trimenon sind notwendig.
Eisenüberladung bei Thalassaemia intermedia
Auch bei der Thalassaemia intermedia, die ja dadurch gekennzeichnet ist, dass kein Bedarf einer regelmäßigen
Transfusionstherapie besteht, kommt es durch die in Verbindung mit der exzessiv gesteigerten Erythropoese enorm
vermehrten Resorption von Eisen aus dem Intestinum zu einer pathologisch bedeutsamen Eisenüberladung.
Eisenbilanzuntersuchungen ergaben eine jährliche Eisenbeladung für erwachsene Patienten mit Thalassaemia intermedia
von 2-5g (50). Die durch eine unbehandelte Siderose bei Patienten mit Thalassaemia intermedia zu beobachtenden
Probleme entsprechen im wesentlichen den auch bei Patienten mit Thalassaemia major oder anderen Patienten mit
Eisenüberladung beobachteten, wenn auch in insgesamt geringerer Häufigkeit (12, 51). Im Vergleich zur Thalassaemia
major weist die Eisenüberladung bei der Thalassaemia intermedia einige Besonderheiten auf. So sind häufig im Verhältnis
zur Lebereisenkonzentration inadäquat niedrig erscheinende Ferritinwerte festzustellen (52). Daraus ergibt sich die
Notwendigkeit regelmäßiger Untersuchungen der Lebereisenkonzentration vor allem bei erwachsenen Patienten, um so
frühzeitig eine bedrohliche Siderose erkennen und behandeln zu können. Dies ist u.a. auch vor dem Hintergrund von
Bedeutung, dass die bei der Thalassaemia intermedia im Gegensatz zur Thalassaemia major sich primär hepatozellulär
manifestierende Siderose (bei Thal. major primär Speicherung in Zellen des RES) ein erhöhtes Risiko der Entwicklung einer
Fibrose birgt. Hervorzuheben ist außerdem, dass das Ausmaß der Siderose nach einer Splenektomie deutlich anzusteigen
scheint (53). Bei den kardialen Störungen sind im Gegensatz zur Thalassaemia major Störungen der Rechtsherzfunktion im
Zusammenhang mit einer pulmonalen Hypertension führend (54). Ein hohes linksventrikuläres Auswurfvolumen in Reaktion
auf eine chronische Gewebshypoxie sowie ein erhöhter pulmonaler Gefäßwiderstand sind dabei wichtige pathogenetische
Faktoren. Bezüglich der Knochenerkrankungen (siehe Thal. major) ist bei der Thalassaemia intermedia die besondere Rolle
der Knochenmarkexpansion hervorzuheben, für deren Behandlung der Einsatz von Hydroxyharnstoff erwogen werden
sollte. Generell weisen nahezu alle Patienten mit Thalassaemia intermedia eine Osteopenie/Osteoporose auf, die wie oben
dargestellt behandelt werden muss.
Die Eiseneliminationstherapie erfolgt meist in Form einer unterschiedlich gestalteten Intervalltherapie, seltener als
Dauertherapie analog zu der bei Thalassaemia major. Generell ist hierbei ein sorgfältiges Abwägen und Prüfen hinsichtlich
des Verhältnisses von Eisenüberladung und Therapieintensität besonders bedeutsam, um Überchelierungen mit
entsprechenden Nebenwirkungsrisiken zu verhindern.
Literatur
1.
Heinrich HC, Gabbe EE, Oppitz KH, Whang DH, Bender-Götze C, Schäfer KH, Schroter W, Pfau AA. Absorption of
inorganic and food iron in children with heterozygous and homozygous ß-Thalassemia. Z-Kinderheilkd 1973; 115(1): 1-22
2.
Kulozik AE. Thalassämien. In: Gadner H, Gaedicke G, Niemeyer C, Ritter J (Hrsg). Pädiatrische Hämatologie und
Onkologie. Springer Heidelberg, 2005
3.
Weatherall DJ, Clegg JB. The Thalassemia Syndromes: 4th Edition. Oxford: Blackwell Scientific Publications, 2001
4.
Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med 2005; 353: 1135-46.
5.
Cooley TB, Lee PA. A series of cases of splenomegaly in children with anemia and peculiar bone changes. Trans Am
Pediatr Soc 1925; 37: 29-30
6.
Olivieri NF. The beta-thalassemias. N Engl J Med 1999; 341: 99-109
7.
Kleihauer E. Anomale Hämoglobine und Thalassämiesyndrome: Grundlagen und Klinik. Unter Mitarbeit v. E. Kohne
u. A.E. Kulozik. Landsberg: Ecomed, 1996
8.
De Maria R, Testa U, Luchetti L, Zeuner A, Stassi G, Pelosi E, Riccioni R, Felli N, Samoggia P, Peschle C. Apoptotic
role of Fas/Fas ligand system in the regulation of erythropoiesis. Blood 1999; 93: 796-803
9.
Mathias LA, Fisher TC, Zeng L, Meiselman HJ, Weinberg KI, Hiti AL, Malik P. Ineffective erythropoiesis in beta-
thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp Hematol 2000; 28: 1343-53
10.
Aydingoz U, Oto A, Cila A. Spinal cord compression due to epidural extramedullary haematopoiesis in thalassaemia:
MRI. 1997; 39: 870-872
11.
Cario H, Wegener M, Debatin KM, Kohne E. Treatment with hydroxyurea in thalassemia intermedia with
paravertebral pseudotumors of extramedullary hematopoiesis. Ann Hematol 2002; 81: 478-82
12.
Camaschella C, Cappellini MD. Thalassemia intermedia. Haematologica 1995; 80: 58-68
13.
Storb RF, Lucarelli G, McSweeney PA, Childs RW. Hematopoietic cell transplantation for benign hematological
disorders and solid tumors. Hematology (Am Soc Hematol Educ Program) 2003: 372-97
14.
Giardina PJ, Grady RW. Chelation therapy in beta-thalassemia: the benefits and limitations of desferrioxamine.
Semin Hematol 1995; 32: 304-312
15.
Fosburg M, Nathan D, Wayne A. Desferrioxamine provocative test: methodology for estimating iron and total iron
binding capacity Blood 1990; 76: 2162
16.
Cario H, Stahnke K, Sander S, Kohne E. Epidemiological situation and treatment of patients with thalassemia major
in Germany: results of the German multicenter beta-thalassemia study. Ann Hematol 2000; 79: 7-12
17.
Aessopos A, Farmakis D, Hatziliami A, Fragodimitri C, Karabatsos F, Joussef J, Mitilineou E, Diamanti-Kandaraki E,
Meletis J, Karagiorga M. Cardiac status in well-treated patients with thalassemia major. Eur J Haematol 2004; 73: 359-366
18.
Vecchio C, Derchi G. Management of cardiac complications in patients with thalassemia major. Semin Hematol 1995;
32: 288-296
19.
Anderson LJ, Holden S, Davis B, Prescott E, Charrier CC, Bunce NH, Firmin DN, Wonke B, Porter J, Walker JM,
Pennell DJ. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart
J 2001; 22: 2171-2179
20.
Anderson LJ, Wonke B, Prescott E, Holden S, Walker JM, Pennell DJ. Comparison of effects of oral deferiprone and
subcutaneous desferrioxamine on myocardial iron concentrations and ventricular function in beta-thalassaemia. Lancet
2002; 360: 516-20
21.
Pennell DJ. T2* magnetic resonance and myocardial iron in thalassemia. Ann N Y Acad Sci 2005; 1054: 373-8
22.
Pennell DJ, Berdoukas V, Karagiorga M, Ladis V, Piga A, Aessopos A, Gotsis ED, Tanner MA, Smith GC, Westwood
MA, Wonke B, Galanello R. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients
with asymptomatic myocardial siderosis. Blood 2005; 107:3738-3744
23.
Kremastinos DT, Tiniakos G, Theodorakis GN, Katritsis DG, Toutouzas PK. Myocarditis in beta-thalassemia major. A
cause of heart failure. Circulation 1995; 91: 66-71
24.
Koerner MM, Tenderich G, Minami K, zu KE, Mannebach H, Kleesiek K, Meyer H, Koerfer R. Heart transplantation
for end-stage heart failure caused by iron overload. Brit J Haematol 1997; 97: 293-296
25.
Olivieri NF, Liu PP, Sher GD, Daly PA, Greig PD, McCusker PJ, Collins AF, Francombe WH, Templeton DM, Butany J.
Brief report: combined liver and heart transplantation for end-stage iron-induced organ failure in an adult with homozygous
beta-thalassemia. N Engl J Med 1994; 330: 1125-1127
26.
Cunningham MJ, Macklin EA, Neufeld EJ, Cohen AR. Complications of beta-thalassemia major in North America.
Blood 2004; 104: 34-9
27.
Prati D, Maggioni M, Milani S, Cerino M, Cianciulli P, Coggi G, Forni GL, Magnano C, Meo A, Gramignoli R, Rebulla
P, Fiorelli G, Cappellini MD. Clinical and histological characterization of liver disease in patients with transfusion-dependent
beta-thalassemia. A multicenter study of 117 cases. Haematologica 2004; 89: 1179-86
28.
Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, Romeo MA, Forni GL,
Gamberini MR, Ghilardi R, Piga A, Cnaan A. Survival and complications in patients with thalassemia major treated with
transfusion and deferoxamine. Haematologica 2004; 89: 1187-93
29.
Lucarelli G, Galimberti M, Giardini C, Polchi P, Angelucci E, Baronciani D, Erer B, Gaziev D. Bone marrow
transplantation in thalassemia. The experience of Pesaro. Ann N Y Acad Sci 1998; 850: 270-5
30.
Perifanis V, Tziomalos K, Tsatra I, Karyda S, Patsiaoura K, Athanassiou-Metaxa M. Prevalence and severity of liver
disease in patients with b thalassemia major. A single-institution fifteen-year experience. Haematologica 2005; 90: 1136-8
31.
Angelucci E, Muretto P, Nicolucci A, Baronciani D, Erer B, Gaziev J, Ripalti M, Sodani P, Tomassoni S, Visani G,
Lucarelli G. Effects of iron overload and hepatitis C virus positivity in determining progression of liver fibrosis in thalassemia
following bone marrow transplantation. Blood 2002; 100:17-21
32.
Kattamis CA, Kattamis AC. Management of thalassemias: growth and development, hormone substitution, vitamin
supplementation, and vaccination. Semin Hematol 1995; 32: 269-279
33.
De Sanctis V, Katz M, Vullo C, Bagni B, Ughi M, Wonke B. Effect of different treatment regimes on linear growth and
final height in beta-thalassaemia major. Clin Endocrinol (Oxf) 1994; 40:791-8
34.
Rodda CP. Body proportions in patients with homozygous beta-thalassaemia. Acta Paediatr Suppl 1994; 406:107-8
35.
Cavallo L, Gurrado R, Zecchino C, Manolo F, DeSanctis V, Cisternino M, CarusoNicoletti M, Galati M. Short-term
therapy with recombinant growth hormone in polytransfused thalassaemia major patients with growth deficiency. J Pediatr
Endocrinol Metab 1998; 11:845-849
36.
De Sanctis V. Growth and Puberty and Its Management in Thalassaemia. Hormone Research 2002; 58: 72-79
37.
Bronspiegel WN, Olivieri NF, Tyler B, Andrews DF, Freedman MH, Holland FJ. Effect of age at the start of iron
chelation therapy on gonadal function in beta-thalassemia major. N Engl J Med 1990; 323: 713-719
38.
Allegra A, Capra M, Cuccia L, Pulejo ML, Raineri L, Corselli F, Traina MC, Giannola C, La Grutta A. Hypogonadism in
beta-thalassemic adolescents: a characteristic pituitary-gonadal impairment. The ineffectiveness of long-term iron chelation
therapy. Gynecol Endocrinol 1990; 4: 181-91
39.
Cario H, Holl RW, Debatin KM, Kohne E. Insulin sensitivity and beta-cell secretion in thalassaemia major with
secondary haemochromatosis: assessment by oral glucose tolerance test. Eur J Pediatr 2003; 162: 139-46
40.
Cavallo-Perin P, Pacini G, Cerutti F, Bessone A, Condo C, Sacchetti L, Piga A, Pagano G. Insulin resistance and
hyperinsulinemia in homozygous beta-thalassemia. Metabolism 1995; 44: 281-6
41.
Mangiagli A, Campisi S, De Sanctis V, Nicoletti MC, Cardinale G, Galati MC, Raiola G, Rigano P, Saviano A. Effects
of acarbose in beta-thalassaemia major patients with normal glucose tolerance and hyperinsulinism. Pediatr Endocrinol Rev
2004; 2 Suppl 2: 272-5
42.
DeSanctis V, Vullo C, Bagni B, Chiccoli L. Hypoparathyroidism in beta-thalassemia major. Clinical and laboratory
observations in 24 patients. Acta Haematol 1992; 88: 105-108
43.
Jensen CE, Tuck SM, Agnew JE, Koneru S, Morris RW, Yardumian A, Prescott E, Hoffbrand AV, Wonke B. High
prevalence of low bone mass in thalassaemia major. Br J Haematol 1998; 103: 911-5
44.
Voskaridou E, Terpos E. New insights into the pathophysiology and management of osteoporosis in patients with
beta thalassaemia. Br J Haematol 2004; 127: 127-39
45.
Wonke B. Bone disease in beta-thalassaemia major. Br J Haematol 1998; 103: 897-901
46.
Voskaridou E, Terpos E, Spina G, Palermos J, Rahemtulla A, Loutradi A, Loukopoulos D. Pamidronate is an effective
treatment for osteoporosis in patients with beta-thalassaemia. Br J Haematol 2003; 123: 730-7
47.
Angastiniotis M, Pavlides N, Aristidou K, Kanakas A, Yerakaris M, Eracleous E, Posporis T. Bone pain in
thalassaemia: Assessment of DEXA and MRI findings. J Pediatr Endocrinol Metab 1998; 11: 779-784
48.
Aessopos A, Karabatsos F, Farmakis D, Katsantoni A, Hatziliami A, Youssef J, Karagiorga M. Pregnancy in patients
with well-treated beta-thalassemia: outcome for mothers and
newborn infants. American Journal of Obstetetrics and
Gynecology 1999; 180: 360-365
49.
Jensen CE, Tuck SM, Wonke B. Fertility in beta-thalassaemia major: a report of 16 pregnancies, preconceptual
evaluation and a review of the literature. Br J Obstetr Gynaecol 1995; 102: 625-629
50
DeSanctis V, Tangerini A, Testa MR, Lauriola AL, Gamberini MR, Cavallini AR, Rigolin F. Final height and endocrine
function in thalassaemia intermedia. J Pediatr Endocrinol Metab 1998; 11: 965-971
51.
Pippard MJ, Callender ST, Finch CA. Ferrioxamine excretion in iron-loaded man. Blood 1982; 60: 288-94
52.
Fiorelli G, Fargion S, Piperno A, Battafarano N, Cappellini MD. Iron metabolism in thalassemia intermedia.
Haematologica 1990; 75 Suppl 5: 89-95
53.
Aessopos A, Farmakis D, Karagiorga M, Voskaridou E, Loutradi A, Hatziliami A, Joussef J, Rombos J, Loukopoulos
D. Cardiac involvement in thalassemia intermedia: a multicenter study. Blood 2001; 97:3411-3416











© www.eiseninfo.de
Thalassämien
Thalassämien bilden eine heterogene Gruppe genetisch
bedingter Erkrankungen, bei denen die Bildung normalen
Hämoglobins auf Grund einer defekten Synthese einer
oder mehrerer Globinketten teilweise oder vollständig
gestört ist (2,3). Von der Synthesestörung können alle
Polypeptidketten der Hämoglobine des Menschen (HbA =
a2b2; HbF = a2g2; HbA2 = a2d2) betroffen sein. Bei
meist autosomal rezessivem Erbgang gibt es
heterozygote und homozygote bzw. gemischt-
heterozygote Formen. Klinisch und hinsichtlich ihrer
Häufigkeit besonders bedeutsam ist die b-Thalassämie.

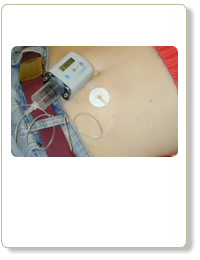
Eisenüberladung
Subkutane Injektion von DesferalR, einem Eisenchelator, der Eisen im Körper mobilisieren und ausscheiden lassen kann