Das Verständnis der sekundären Eisenüberladung wurde wesentlich durch die Erfahrungen mit der
Mittelmeeranämie (β-Thalassämie, Morbus Cooley) gewonnen. Deswegen soll im Folgenden die Situation bei
der β-Thalassaemia major beschrieben werden. Die meisten Patienten mit β-Thalassaemia major werden
zwischen dem 6. Lebensmonat und dem 2. Lebensjahr mit einer Anämie diagnostiziert, wenn die Umstellung
von fetalem Hämoglobin F (α2γ2) auf adultes Hämoglobin A (α2β2) erfolgt ist. Nach Diagnosestellung wird mit der
Transfusionsbehandlung begonnen. Entscheidend für die Behandlung der Thalassämie mit Transfusionen ist
die Balance zwischen Eisenbelastung und einer wirksamen Suppression der ineffektiven Erythropoese.
Hämolyse und ineffektive Erythropoese verursachen die Anämie, der jeweilige Anteil variiert bei den
verschiedenen Formen der Thalassämie. Das Knochenmark von Patienten mit β-Thalassämie enthält etwa 5-6
mal so viel erythropoetische Vorläuferzellen wie das Knochenmark gesunder Personen und etwa 15 mal so
viele apoptotische Zellen (2). Die beschleunigte Apoptose wird durch die Ablagerung freier alpha-Globinketten
verursacht (3). Eine zu geringe Transfusionsfrequenz oder –menge führt zu einer Expansion der medullären
und extramedullären Blutbildung. Die Folgen sind Knochendeformitäten mit typischen Veränderungen wie
Bürstenschädel, prominenten Stirnhöckern und Kieferknochen, Prognathie, Ausdünnung der Kompakta und
Hepatosplenomegalie (4-6). Regelmäßige Transfusionen verhindern dies und verbessern Wachstum und
Entwicklung (7).
Zudem führt die ineffektive, gesteigerte Erythropoese zur vermehrten intestinalen Eisenaufnahme. Zurzeit gilt
ein Transfusionsregime, welches einen Hämoglobinwert von 9-10 g/dl prätransfusionem anstrebt als ideal. Mit
diesem Regime wachsen Kinder besser, Hepatosplenomegalie und Knochendeformitäten sind geringer
ausgeprägt (8).
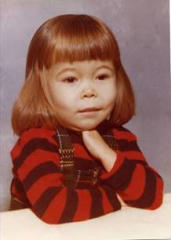
Abb. 1. Links: Bild eines 4-Jahre alten Mädchens mit unbehandelter ß-Thalassaemia major. Typischer
Gesichtsschädel mit prominenter Maxilla und verbreiteter Nasenwurzel. Rechts: Gleiche Patientin im Alter von
29 Jahren (mit Ihren beiden Kindern). Transfusionstherapie ab dem 4.5 Lebensjahr. Chelattherapie mit ca. 7
Jahren, seither optimale Behandlung mit DFO.
Erreicht wird dies bei den meisten Patienten mit Thalassaemia major durch 15-20ml/kg Erythrozytenkonzentrat in 3-4-wöchentlichem Abstand (Tabelle 1). In der Vergangenheit wurden Hypertransfusions-Schemata
praktiziert, bei denen schon bei einem Hb-Wert unter 12 g/dl eine erneute Transfusion durchgeführt wurde (9).
Solche Schemata führten zu einer raschen Eisenüberladung, die oft nur schwierig durch Deferoxamin zu
kontrollieren war. Auf der anderen Seite sollte man gerade bei Kindern den Hb-Wert nicht zu weit abfallen
lassen, um keine Hirnreifungsstörungen zu riskieren.
Transfusionstherapie bei ß-Thalassämie
Therapiebeginn
Hb < 8 g/dl
Basis-Hämoglobin
> 9-10 g/dl
Transfusionsintervall
3-4 Wochen
Transfusionsmenge
15-20 ml/kg
(Erythrozytenkonzentrat 60 %)
Tab. 1: Richtgrößen bei der Transfusionstherapie bei ß-Thalassämie.
Die Transfusionsgeschwindigkeit muss dem klinischen Zustand des Patienten angepasst werden. Bei neu
diagnostizierten Patienten mit länger bestehender schwerer Anämie sollte eine vorsichtige und langsame
Transfusion durchgeführt werden, um eine kardiale Belastung zu verhindern.
Das Hauptrisiko der lebenslangen Transfusionstherapie besteht in der Eisenüberladung. Darüber hinaus kann
es zur Alloimmunisierung kommen und Antikörper-Suchtests sollten einmal pro Jahr angefordert werden. Einige
Behandlungszentren empfehlen deshalb die Verwendung von Präparaten, die bezüglich aller Rhesus-Antigene
identisch sind. Solange bei einem CMV-negativen Patienten mit Thalassämie die Option für eine
Stammzelltransplantation gegeben ist, sollten CMV-negative Erythrozytenkonzentrate transfundiert werden, um
das Risiko der CMV-Reaktivierung nach Transplantation zu minimieren. Viele Patienten mit Thalassämie
entwickeln im Laufe der Zeit eine ausgeprägte Splenomegalie. Der damit verbundene Hypersplenismus führt zu
einem gesteigerten Transfusionsbedarf. Die Menge an verabreichten Erythrozytenkonzentraten in ml bzw. g
sollte dokumentiert werden, bei einem Verbrauch über 250 ml/kg x Jahr kann eine Splenektomie indiziert sein
(10).
Nach vielen Jahren regelmäßiger Transfusionen gibt es bei einigen Patienten das Problem des venösen
Zugangs. Gelegentlich wird dann die Implantation eines Port-a-Cath-Katheters notwendig. Die Hauptprobleme
sind hier die Katheterinfektion und –obstruktion. Erstere lässt sich durch Vancomycin bzw. Teicoplanin
behandeln, bei letzterer ist die Applikation von 5000-10000 E Urokinase in das Volumen des Porth-Systems
wirksam. Wenn Ausbildung und Beruf geringe Fehlzeiten erforderlich machen, können die Transfusionen auch
übernacht und am Wochenende verabreicht werden.
Vor der Ära der Transfusionstherapie betrug die Lebenserwartung von Kindern mit homozygoter ß-Thalassämie
oft nur wenige Jahre. Es kam zu schweren Knochendeformitäten, massiven Hepatosplenomegalien und Tod
durch Herzversagen. Mit regelmäßigen Transfusionen kann nicht nur die Anämie behandelt werden, sondern es
läßt sich auch den orthopädischen Problemen vorbeugen. Allerdings kommt es ohne Chelatortherapie zu
endokrinen Folgeschäden, Leberfibrose und –zirrhose und die meisten Patienten versterben in der zweiten
Lebensdekade an einer Herzinsuffizienz infolge der kardialen Hämosiderose (11).
Literatur
1.
Use of blood products for elective surgery in 43 European hospitals. The Sanguis Study Group. Transfus Med 1994;
4:251-268
2.
Centis F, Tabellini L, Lucarelli G, Buffi O, Tonucci P, Persini B, Annibali M, Emiliani R, Iliescu A, Rapa S, Rossi R, Ma,
Angelucci E, and Schrier SL. The importance of erythroid expansion in determining the extent of apoptosis in
erythroid precursors in patients with ß-thalassemia major. Blood 2000; 96:3624-3629
3.
Pootrakul P, Sirankapracha P, Hemsorach S. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid
precursor apoptosis in Thai patients with thalassemia. Blood 2000; 96:2606-2612
4.
Caffey J. Cooley’s anemia: a review of the roentgenographic findings in the skeleton: Hickey lecture, 1957. Am J
Roentgenol Radium Ther Nucl Med 1957; 78: 381-391
5.
Choremis C, Liakakos D, Tseghi C, Moschovakis C. Pathogenesis of osseous lesions in thalassemia. J Pediatr 1965;
66:962-963
6.
Orkin SH, Nathan DG. The Thalassemias. In: Nathan and Oski’s Hematology of infancy and childhood. Nathan DG,
Orkin SH, Ginsburg D, Look AT (Eds.), 6th Ed., Saunders, 2003; 843-919
7.
Cunningham MJ, Macklin EA, Neufeld EJ, Cohen AR. Complications of ß-thalassemia major in North America. Blood
2004; 104:34-39
8.
Old JM, Olivieri NF, Thein SL. Diagnosis and management of thalassaemia. In: Weatherall DJ, Clegg B, eds. The
thalassaemia syndromes. 4th ed. Oxford, England: Blackwell Science, 2001; 630-685
9.
Cazzola M, De Stefano P, Ponchio L, Locatelli F, Beguin Y, Dessi C, Barella S, Cao A, Galanello R. Relationship
between transfusion regimen and suppression of erythropoiesis in beta-thalassaemia major. Br J Haematol 1995;
89:473-478
10.
Graziano JH, Piomelli S, Hilgartner M, Giardina P, Karpatkin M, Andrew M, Lolacono N, Seaman C. Chelation
therapy in thalassemia major. III. The role of splenectomy in achieving iron balance. J Pediatr 1981; 99:695-699
11.
Nathan DG. Thalassemia: The continued challenge. Ann NY Acad Sci 2005; 1054:1-
10
in 3-4-wöchentlichem Abstand (Tabelle 1). In der Vergangenheit wurden Hypertransfusions-Schemata
praktiziert, bei denen schon bei einem Hb-Wert unter 12 g/dl eine erneute Transfusion durchgeführt wurde (9).
Solche Schemata führten zu einer raschen Eisenüberladung, die oft nur schwierig durch Deferoxamin zu
kontrollieren war. Auf der anderen Seite sollte man gerade bei Kindern den Hb-Wert nicht zu weit abfallen
lassen, um keine Hirnreifungsstörungen zu riskieren.
Transfusionstherapie bei ß-Thalassämie
Therapiebeginn
Hb < 8 g/dl
Basis-Hämoglobin
> 9-10 g/dl
Transfusionsintervall
3-4 Wochen
Transfusionsmenge
15-20 ml/kg
(Erythrozytenkonzentrat 60 %)
Tab. 1: Richtgrößen bei der Transfusionstherapie bei ß-Thalassämie.
Die Transfusionsgeschwindigkeit muss dem klinischen Zustand des Patienten angepasst werden. Bei neu
diagnostizierten Patienten mit länger bestehender schwerer Anämie sollte eine vorsichtige und langsame
Transfusion durchgeführt werden, um eine kardiale Belastung zu verhindern.
Das Hauptrisiko der lebenslangen Transfusionstherapie besteht in der Eisenüberladung. Darüber hinaus kann
es zur Alloimmunisierung kommen und Antikörper-Suchtests sollten einmal pro Jahr angefordert werden. Einige
Behandlungszentren empfehlen deshalb die Verwendung von Präparaten, die bezüglich aller Rhesus-Antigene
identisch sind. Solange bei einem CMV-negativen Patienten mit Thalassämie die Option für eine
Stammzelltransplantation gegeben ist, sollten CMV-negative Erythrozytenkonzentrate transfundiert werden, um
das Risiko der CMV-Reaktivierung nach Transplantation zu minimieren. Viele Patienten mit Thalassämie
entwickeln im Laufe der Zeit eine ausgeprägte Splenomegalie. Der damit verbundene Hypersplenismus führt zu
einem gesteigerten Transfusionsbedarf. Die Menge an verabreichten Erythrozytenkonzentraten in ml bzw. g
sollte dokumentiert werden, bei einem Verbrauch über 250 ml/kg x Jahr kann eine Splenektomie indiziert sein
(10).
Nach vielen Jahren regelmäßiger Transfusionen gibt es bei einigen Patienten das Problem des venösen
Zugangs. Gelegentlich wird dann die Implantation eines Port-a-Cath-Katheters notwendig. Die Hauptprobleme
sind hier die Katheterinfektion und –obstruktion. Erstere lässt sich durch Vancomycin bzw. Teicoplanin
behandeln, bei letzterer ist die Applikation von 5000-10000 E Urokinase in das Volumen des Porth-Systems
wirksam. Wenn Ausbildung und Beruf geringe Fehlzeiten erforderlich machen, können die Transfusionen auch
übernacht und am Wochenende verabreicht werden.
Vor der Ära der Transfusionstherapie betrug die Lebenserwartung von Kindern mit homozygoter ß-Thalassämie
oft nur wenige Jahre. Es kam zu schweren Knochendeformitäten, massiven Hepatosplenomegalien und Tod
durch Herzversagen. Mit regelmäßigen Transfusionen kann nicht nur die Anämie behandelt werden, sondern es
läßt sich auch den orthopädischen Problemen vorbeugen. Allerdings kommt es ohne Chelatortherapie zu
endokrinen Folgeschäden, Leberfibrose und –zirrhose und die meisten Patienten versterben in der zweiten
Lebensdekade an einer Herzinsuffizienz infolge der kardialen Hämosiderose (11).
Literatur
1.
Use of blood products for elective surgery in 43 European hospitals. The Sanguis Study Group. Transfus Med 1994;
4:251-268
2.
Centis F, Tabellini L, Lucarelli G, Buffi O, Tonucci P, Persini B, Annibali M, Emiliani R, Iliescu A, Rapa S, Rossi R, Ma,
Angelucci E, and Schrier SL. The importance of erythroid expansion in determining the extent of apoptosis in
erythroid precursors in patients with ß-thalassemia major. Blood 2000; 96:3624-3629
3.
Pootrakul P, Sirankapracha P, Hemsorach S. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid
precursor apoptosis in Thai patients with thalassemia. Blood 2000; 96:2606-2612
4.
Caffey J. Cooley’s anemia: a review of the roentgenographic findings in the skeleton: Hickey lecture, 1957. Am J
Roentgenol Radium Ther Nucl Med 1957; 78: 381-391
5.
Choremis C, Liakakos D, Tseghi C, Moschovakis C. Pathogenesis of osseous lesions in thalassemia. J Pediatr 1965;
66:962-963
6.
Orkin SH, Nathan DG. The Thalassemias. In: Nathan and Oski’s Hematology of infancy and childhood. Nathan DG,
Orkin SH, Ginsburg D, Look AT (Eds.), 6th Ed., Saunders, 2003; 843-919
7.
Cunningham MJ, Macklin EA, Neufeld EJ, Cohen AR. Complications of ß-thalassemia major in North America. Blood
2004; 104:34-39
8.
Old JM, Olivieri NF, Thein SL. Diagnosis and management of thalassaemia. In: Weatherall DJ, Clegg B, eds. The
thalassaemia syndromes. 4th ed. Oxford, England: Blackwell Science, 2001; 630-685
9.
Cazzola M, De Stefano P, Ponchio L, Locatelli F, Beguin Y, Dessi C, Barella S, Cao A, Galanello R. Relationship
between transfusion regimen and suppression of erythropoiesis in beta-thalassaemia major. Br J Haematol 1995;
89:473-478
10.
Graziano JH, Piomelli S, Hilgartner M, Giardina P, Karpatkin M, Andrew M, Lolacono N, Seaman C. Chelation
therapy in thalassemia major. III. The role of splenectomy in achieving iron balance. J Pediatr 1981; 99:695-699
11.
Nathan DG. Thalassemia: The continued challenge. Ann NY Acad Sci 2005; 1054:1-
10
in 3-4-wöchentlichem Abstand (Tabelle 1). In der Vergangenheit wurden Hypertransfusions-Schemata
praktiziert, bei denen schon bei einem Hb-Wert unter 12 g/dl eine erneute Transfusion durchgeführt wurde (9).
Solche Schemata führten zu einer raschen Eisenüberladung, die oft nur schwierig durch Deferoxamin zu
kontrollieren war. Auf der anderen Seite sollte man gerade bei Kindern den Hb-Wert nicht zu weit abfallen
lassen, um keine Hirnreifungsstörungen zu riskieren.
Transfusionstherapie bei ß-Thalassämie
Therapiebeginn
Hb < 8 g/dl
Basis-Hämoglobin
> 9-10 g/dl
Transfusionsintervall
3-4 Wochen
Transfusionsmenge
15-20 ml/kg
(Erythrozytenkonzentrat 60 %)
Tab. 1: Richtgrößen bei der Transfusionstherapie bei ß-Thalassämie.
Die Transfusionsgeschwindigkeit muss dem klinischen Zustand des Patienten angepasst werden. Bei neu
diagnostizierten Patienten mit länger bestehender schwerer Anämie sollte eine vorsichtige und langsame
Transfusion durchgeführt werden, um eine kardiale Belastung zu verhindern.
Das Hauptrisiko der lebenslangen Transfusionstherapie besteht in der Eisenüberladung. Darüber hinaus kann
es zur Alloimmunisierung kommen und Antikörper-Suchtests sollten einmal pro Jahr angefordert werden. Einige
Behandlungszentren empfehlen deshalb die Verwendung von Präparaten, die bezüglich aller Rhesus-Antigene
identisch sind. Solange bei einem CMV-negativen Patienten mit Thalassämie die Option für eine
Stammzelltransplantation gegeben ist, sollten CMV-negative Erythrozytenkonzentrate transfundiert werden, um
das Risiko der CMV-Reaktivierung nach Transplantation zu minimieren. Viele Patienten mit Thalassämie
entwickeln im Laufe der Zeit eine ausgeprägte Splenomegalie. Der damit verbundene Hypersplenismus führt zu
einem gesteigerten Transfusionsbedarf. Die Menge an verabreichten Erythrozytenkonzentraten in ml bzw. g
sollte dokumentiert werden, bei einem Verbrauch über 250 ml/kg x Jahr kann eine Splenektomie indiziert sein
(10).
Nach vielen Jahren regelmäßiger Transfusionen gibt es bei einigen Patienten das Problem des venösen
Zugangs. Gelegentlich wird dann die Implantation eines Port-a-Cath-Katheters notwendig. Die Hauptprobleme
sind hier die Katheterinfektion und –obstruktion. Erstere lässt sich durch Vancomycin bzw. Teicoplanin
behandeln, bei letzterer ist die Applikation von 5000-10000 E Urokinase in das Volumen des Porth-Systems
wirksam. Wenn Ausbildung und Beruf geringe Fehlzeiten erforderlich machen, können die Transfusionen auch
übernacht und am Wochenende verabreicht werden.
Vor der Ära der Transfusionstherapie betrug die Lebenserwartung von Kindern mit homozygoter ß-Thalassämie
oft nur wenige Jahre. Es kam zu schweren Knochendeformitäten, massiven Hepatosplenomegalien und Tod
durch Herzversagen. Mit regelmäßigen Transfusionen kann nicht nur die Anämie behandelt werden, sondern es
läßt sich auch den orthopädischen Problemen vorbeugen. Allerdings kommt es ohne Chelatortherapie zu
endokrinen Folgeschäden, Leberfibrose und –zirrhose und die meisten Patienten versterben in der zweiten
Lebensdekade an einer Herzinsuffizienz infolge der kardialen Hämosiderose (11).
Literatur
1.
Use of blood products for elective surgery in 43 European hospitals. The Sanguis Study Group. Transfus Med 1994;
4:251-268
2.
Centis F, Tabellini L, Lucarelli G, Buffi O, Tonucci P, Persini B, Annibali M, Emiliani R, Iliescu A, Rapa S, Rossi R, Ma,
Angelucci E, and Schrier SL. The importance of erythroid expansion in determining the extent of apoptosis in
erythroid precursors in patients with ß-thalassemia major. Blood 2000; 96:3624-3629
3.
Pootrakul P, Sirankapracha P, Hemsorach S. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid
precursor apoptosis in Thai patients with thalassemia. Blood 2000; 96:2606-2612
4.
Caffey J. Cooley’s anemia: a review of the roentgenographic findings in the skeleton: Hickey lecture, 1957. Am J
Roentgenol Radium Ther Nucl Med 1957; 78: 381-391
5.
Choremis C, Liakakos D, Tseghi C, Moschovakis C. Pathogenesis of osseous lesions in thalassemia. J Pediatr 1965;
66:962-963
6.
Orkin SH, Nathan DG. The Thalassemias. In: Nathan and Oski’s Hematology of infancy and childhood. Nathan DG,
Orkin SH, Ginsburg D, Look AT (Eds.), 6th Ed., Saunders, 2003; 843-919
7.
Cunningham MJ, Macklin EA, Neufeld EJ, Cohen AR. Complications of ß-thalassemia major in North America. Blood
2004; 104:34-39
8.
Old JM, Olivieri NF, Thein SL. Diagnosis and management of thalassaemia. In: Weatherall DJ, Clegg B, eds. The
thalassaemia syndromes. 4th ed. Oxford, England: Blackwell Science, 2001; 630-685
9.
Cazzola M, De Stefano P, Ponchio L, Locatelli F, Beguin Y, Dessi C, Barella S, Cao A, Galanello R. Relationship
between transfusion regimen and suppression of erythropoiesis in beta-thalassaemia major. Br J Haematol 1995;
89:473-478
10.
Graziano JH, Piomelli S, Hilgartner M, Giardina P, Karpatkin M, Andrew M, Lolacono N, Seaman C. Chelation
therapy in thalassemia major. III. The role of splenectomy in achieving iron balance. J Pediatr 1981; 99:695-699
11.
Nathan DG. Thalassemia: The continued challenge. Ann NY Acad Sci 2005; 1054:1-
10













© www.eiseninfo.de
Transfusionstherapie
Die Bluttransfusion ist bei kritischem Blutverlust oder bei
lebensbedrohlichem Mangel an Blutbestandteilen nach
wie vor eine lebensrettende und unverzichtbare Methode
der Medizin. Als allgemeiner Grenzwert für die gegebene
Indikation für einen Bluttransfusion gilt in Deutschland
seit vielen Jahrzehnten ein Hämoglobinwert von 10 g/dl,
obwohl die von der EU-Kommision beauftragte
SANGUIS-Studie (1994) diesen Wert als zu hoch
einschätzt (1).
In dem hier diskutierten Zusammenhang wird eine
chronische Transfusionstherapie bei Blutkrankheiten in
dem Sinne einer Blutbildungsstörung (Anämien,
hämatologische Neoplasien, Aplastische Anämien)
eingesetzt.
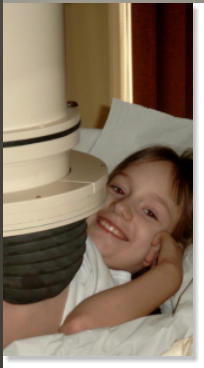
Eisenchelatortherapie
Subkutane Therapie mit Desferal mit einer kleinen Pumpe meist über Nacht.